Unit 4 Chemical Kinetics
Chemistry, by its very nature, is concerned with change. Substances with well defined properties are converted by chemical reactions into other substances with different properties. For any chemical reaction, chemists try to find out
(a) the feasibility of a chemical reaction which can be predicted by thermodynamics ( as you know that a reaction with DG < 0, at constant temperature and pressure is feasible);
(b) extent to which a reaction will proceed can be determined from chemical equilibrium;
(c) speed of a reaction i.e. time taken by a reaction to reach equilibrium.
Along with feasibility and extent, it is equally important to know the rate and the factors controlling the rate of a chemical reaction for its complete understanding. For example, which parameters determine as to how rapidly food gets spoiled? How to design a rapidly setting material for dental filling? Or what controls the rate at which fuel burns in an auto engine? All these questions can be answered by the branch of chemistry, which deals with the study of reaction rates and their mechanisms, called chemical kinetics. The word kinetics is derived from the Greek word ‘kinesis’ meaning movement. Thermodynamics tells only about the feasibility of a reaction whereas chemical kinetics tells about the rate of a reaction. For example, thermodynamic data indicate that diamond shall convert to graphite but in reality the conversion rate is so slow that the change is not perceptible at all. Therefore, most people think that diamond is forever. Kinetic studies not only help us to determine the speed or rate of a chemical reaction but also describe the conditions by which the reaction rates can be altered. The factors such as concentration, temperature, pressure and catalyst affect the rate of a reaction. At the macroscopic level, we are interested in amounts reacted or formed and the rates of their consumption or formation. At the molecular level, the reaction mechanisms involving orientation and energy of molecules undergoing collisions, are discussed.
In this Unit, we shall be dealing with average and instantaneous rate of reaction and the factors affecting these. Some elementary ideas about the collision theory of reaction rates are also given. However, in order to understand all these, let us first learn about the reaction rate.
4.1 Rate of a Chemical Reaction
Some reactions such as ionic reactions occur very fast, for example, precipitation of silver chloride occurs instantaneously by mixing of aqueous solutions of silver nitrate and sodium chloride. On the other hand, some reactions are very slow, for example, rusting of iron in the presence of air and moisture. Also there are reactions like inversion of cane sugar and hydrolysis of starch, which proceed with a moderate speed. Can you think of more examples from each category?
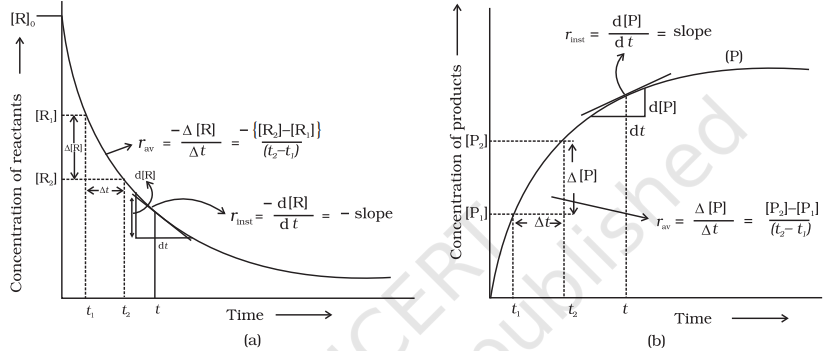
You must be knowing that speed of an automobile is expressed in terms of change in the position or distance covered by it in a certain period of time. Similarly, the speed of a reaction or the rate of a reaction can be defined as the change in concentration of a reactant or product in unit time. To be more specific, it can be expressed in terms of:
(i) the rate of decrease in concentration of any one of the reactants, or
(ii) the rate of increase in concentration of any one of the products. Consider a hypothetical reaction, assuming that the volume of the system remains constant.
$ \mathrm{R} \rightarrow \mathrm{P} $ One mole of the reactant $R$ produces one mole of the product $P$. If $\left[R\right]_1$ and $\left[P\right]_1$ are the concentrations of $R$ and $P$ respectively at time $t_1$ and $[\mathrm{R}]_2$ and $[\mathrm{P}]_2$ are their concentrations at time $\mathrm{t_2}$ then,
$$ \begin{aligned} \Delta t & =t_{2}-t_1 \\ \Delta[\mathrm{R}] & =[\mathrm{R}]_2-[\mathrm{R}]_1 \\ \Delta[\mathrm{P}] & =[\mathrm{P}]_2-[\mathrm{P}]_1 \end{aligned} $$
The square brackets in the above expressions are used to express molar concentration.
Rate of disappearance of $R$
$$ \begin{equation*} =\frac{\text { Decrease in concentration of } \mathrm{R}}{\text { Time taken }}=-\frac{\Delta[\mathrm{R}]}{\Delta t} \tag{4.1} \end{equation*} $$
Rate of appearance of $\mathrm{P}$
$$ \begin{equation*} =\frac{\text { Increase in concentration of } \mathrm{P}}{\text { Time taken }}=+\frac{\Delta[\mathrm{P}]}{\Delta t} \tag{4.2} \end{equation*} $$
Since, $\Delta[R]$ is a negative quantity (as concentration of reactants is decreasing), it is multiplied with -1 to make the rate of the reaction a positive quantity.
Equations (4.1) and (4.2) given above represent the average rate of a reaction, $r_{\mathrm{av}}$.
Average rate depends upon the change in concentration of reactants or products and the time taken for that change to occur (Fig. 4.1).
Units of rate of a reaction
From equations (3.1) and (3.2), it is clear that units of rate are concentration time ${ }^{-1}$. For example, if concentration is in $\mathrm{mol} \mathrm{L}^{-1}$ and time is in seconds then the units will be $\mathrm{mol} \mathrm{L}^{-1} \mathrm{~s}^{-1}$. However, in gaseous reactions, when the concentration of gases is expressed in terms of their partial pressures, then the units of the rate equation will be atm $\mathrm{s}^{-1}$.
4.2 Factors Influencing Rate of a Reaction
Rate of reaction depends upon the experimental conditions such as concentration of reactants (pressure in case of gases), temperature and catalyst.
4.2.1 Dependence of Rate on Concentration
The rate of a chemical reaction at a given temperature may depend on the concentration of one or more reactants and products. The representation of rate of reaction in terms of concentration of the reactants is known as rate law. It is also called as rate equation or rate expression.
4.2.2 Rate Expression and Rate Constant
The results in Table 4.1 clearly show that rate of a reaction decreases with the passage of time as the concentration of reactants decrease. Conversely, rates generally increase when reactant concentrations increase. So, rate of a reaction depends upon the concentration of reactants.
$$ \mathrm{aA}+\mathrm{bB} \rightarrow \mathrm{cC}+\mathrm{dD} $$
where a, b, c and d are the stoichiometric coefficients of reactants and products.
The rate expression for this reaction is
$$ \begin{equation*} \text { Rate } \propto[\mathrm{A}]^{\mathrm{x}}[\mathrm{B}]^{\mathrm{y}} \tag{4.4} \end{equation*} $$
where exponents $\mathrm{x}$ and $\mathrm{y}$ may or may not be equal to the stoichiometric coefficients ( $\mathrm{a}$ and $\mathrm{b}$ ) of the reactants. Above equation can also be written as
$$ \begin{align*} & \text { Rate }=k[\mathrm{~A}]^{\mathrm{x}} \quad[\mathrm{B}]^{\mathrm{y}} \tag{4.4a}\\ & -\frac{\mathrm{d}[\mathrm{R}]}{\mathrm{d} t}=k[\mathrm{~A}]^{\mathrm{x}}[\mathrm{B}]^{\mathrm{y}} \tag{4.4b} \end{align*} $$
This form of equation (4.4 b) is known as differential rate equation, where k is a proportionality constant called rate constant. The equation like (4.4), which relates the rate of a reaction to concentration of reactants is called rate law or rate expression. Thus, rate law is the expression in which reaction rate is given in terms of molar concentration of reactants with each term raised to some power, which may or may not be same as the stoichiometric coefficient of the reacting species in a balanced chemical equation. For example:
$$ 2 \mathrm{NO}(\mathrm{g})+\mathrm{O_2}(\mathrm{~g}) \rightarrow 2 \mathrm{NO_2}(\mathrm{~g}) $$
We can measure the rate of this reaction as a function of initial concentrations either by keeping the concentration of one of the reactants constant and changing the concentration of the other reactant or by changing the concentration of both the reactants. The following results are obtained (Table 4.2).

It is obvious, after looking at the results, that when the concentration of $\mathrm{NO}$ is doubled and that of $\mathrm{O_2}$ is kept constant then the initial rate increases by a factor of four from 0.096 to $0.384 \mathrm{~mol} \mathrm{~L}^{-1} \mathrm{~s}^{-1}$. This indicates that the rate depends upon the square of the concentration of NO. When concentration of NO is kept constant and concentration of $\mathrm{O_2}$ is doubled the rate also gets doubled indicating that rate depends on concentration of $\mathrm{O_2}$ to the first power. Hence, the rate equation for this reaction will be
$$ \text { Rate }=k\left[\mathrm{NO}^{2}\left[\mathrm{O_2}\right]\right]. $$
The differential form of this rate expression is given as
$$ -\frac{\mathrm{d}[\mathrm{R}]}{\mathrm{d} t}=k[\mathrm{NO}]^{2}\left[\mathrm{O_2}\right] $$
Now, we observe that for this reaction in the rate equation derived from the experimental data, the exponents of the concentration terms are the same as their stoichiometric coefficients in the balanced chemical equation.
Some other examples are given below: Reaction Experimental rate expression
1. $\mathrm{CHCl_3}+\mathrm{Cl_2} \rightarrow \mathrm{CCl_4}+\mathrm{HCl}$
- Rate $=k\left[\mathrm{CHC_3}\right]\left[\mathrm{Cl_2}\right]$
2. $\mathrm{CH_3} \mathrm{COOC_2} \mathrm{H_5}+\mathrm{H_2} \mathrm{O} \rightarrow \mathrm{CH_3} \mathrm{COOH}+\mathrm{C_2} \mathrm{H_5} \mathrm{OH}$ Rate $=k\left[\mathrm{CH_3} \mathrm{COOC_2} \mathrm{H_5}\right]^{1}\left[\mathrm{H_2} \mathrm{O}\right]^{0}$
In these reactions, the exponents of the concentration terms are not the same as their stoichiometric coefficients. Thus, we can say that:
Rate law for any reaction cannot be predicted by merely looking at the balanced chemical equation, i.e., theoretically but must be determined experimentally.
4.2.3 Order of a Reaction
In the rate equation (4.4)
Reaction $\quad$ Rate $=k[A]^{\mathrm{x}}[\mathrm{B}]^{\mathrm{y}}$
$\mathrm{x}$ and $\mathrm{y}$ indicate how sensitive the rate is to the change in concentration of A and B. Sum of these exponents, i.e., $x+y$ in (4.4) gives the overall order of a reaction whereas $\mathrm{x}$ and $\mathrm{y}$ represent the order with respect to the reactants $\mathrm{A}$ and $\mathrm{B}$ respectively.
Hence, the sum of powers of the concentration of the reactants in the rate law expression is called the order of that chemical reaction.
Order of a reaction can be $0,1,2,3$ and even a fraction. A zero order reaction means that the rate of reaction is independent of the concentration of reactants.
4.2.4 Molecularity of a Reaction
Another property of a reaction called molecularity helps in understanding its mechanism. The number of reacting species (atoms, ions or molecules) taking part in an elementary reaction, which must collide simultaneously in order to bring about a chemical reaction is called molecularity of a reaction. The reaction can be unimolecular when one reacting species is involved, for example, decomposition of ammonium nitrite.
$ \mathrm{NH_4} \mathrm{NO_2} \rightarrow \mathrm{N_2}+2 \mathrm{H_2} \mathrm{O} $ Bimolecular reactions involve simultaneous collision between two species, for example, dissociation of hydrogen iodide.
$ 2 \mathrm{HI} \rightarrow \mathrm{H_2}+\mathrm{I_2} $ Trimolecular or termolecular reactions involve simultaneous collision between three reacting species, for example, $2 \mathrm{NO}+\mathrm{O_2} \rightarrow 2 \mathrm{NO_2}$
The probability that more than three molecules can collide and react simultaneously is very small. Hence, reactions with the molecularity three are very rare and slow to proceed.
It is, therefore, evident that complex reactions involving more than three molecules in the stoichiometric equation must take place in more than one step.
$$ \mathrm{KClO_3}+6 \mathrm{FeSO_4}+3 \mathrm{H_2} \mathrm{SO_4} \rightarrow \mathrm{KCl}+3 \mathrm{Fe_2}\left(\mathrm{SO_4}\right)_{3}+3 \mathrm{H_2} \mathrm{O} $$
This reaction which apparently seems to be of tenth order is actually a second order reaction. This shows that this reaction takes place in several steps. Which step controls the rate of the overall reaction? The question can be answered if we go through the mechanism of reaction, for example, chances to win the relay race competition by a team depend upon the slowest person in the team. Similarly, the overall rate of the reaction is controlled by the slowest step in a reaction called the rate determining step. Consider the decomposition of hydrogen peroxide which is catalysed by iodide ion in an alkaline medium.
$$ 2 \mathrm{H_2} \mathrm{O_2} \xrightarrow[\text { Alkaline medium }]{\mathrm{I}^{-}} 2 \mathrm{H_2} \mathrm{O}+\mathrm{O_2} $$
The rate equation for this reaction is found to be
$$ \text { Rate }=\frac{-\mathrm{d}\left[\mathrm{H_2} \mathrm{O_2}\right]}{\mathrm{d} t}=k\left[\mathrm{H_2} \mathrm{O_2}\right]\left[\mathrm{I}^{-}\right] $$
This reaction is first order with respect to both $\mathrm{H_2} \mathrm{O_2}$ and $\mathrm{I}^{-}$. Evidences suggest that this reaction takes place in two steps
(1) $\mathrm{H_2} \mathrm{O_2}+\mathrm{I}^{-} \rightarrow \mathrm{H_2} \mathrm{O}+\mathrm{IO}^{-}$
(2) $\mathrm{H_2} \mathrm{O_2}+\mathrm{IO}^{-} \rightarrow \mathrm{H_2} \mathrm{O}+\mathrm{I}^{-}+\mathrm{O_2}$
Both the steps are bimolecular elementary reactions. Species IO- is called as an intermediate since it is formed during the course of the reaction but not in the overall balanced equation. The first step, being slow, is the rate determining step. Thus, the rate of formation of intermediate will determine the rate of this reaction.
Thus, from the discussion, till now, we conclude the following:
(i) Order of a reaction is an experimental quantity. It can be zero and even a fraction but molecularity cannot be zero or a non integer.
(ii) Order is applicable to elementary as well as complex reactions whereas molecularity is applicable only for elementary reactions. For complex reaction molecularity has no meaning.
(iii) For complex reaction, order is given by the slowest step and molecularity of the slowest step is same as the order of the overall reaction.
4.3 Integrated Rate Equations
We have already noted that the concentration dependence of rate is called differential rate equation. It is not always convenient to determine the instantaneous rate, as it is measured by determination of slope of the tangent at point ‘t’ in concentration vs time plot (Fig. 4.1). This makes it difficult to determine the rate law and hence the order of the reaction. In order to avoid this difficulty, we can integrate the differential rate equation to give a relation between directly measured experimental data, i.e., concentrations at different times and rate constant.
The integrated rate equations are different for the reactions of different reaction orders. We shall determine these equations only for zero and first order chemical reactions.
4.3.1 Zero Order Reactions
Zero order reaction means that the rate of the reaction is proportional to zero power of the concentration of reactants. Consider the reaction, $$ \begin{aligned} & \mathrm{R} \rightarrow \mathrm{P} \ & \text { Rate }=-\frac{\mathrm{d}[\mathrm{R}]}{\mathrm{d} t}=k[\mathrm{R}]^{0} \end{aligned} $$
As any quantity raised to power zero is unity
$$ \begin{aligned} & \text { Rate }=-\frac{\mathrm{d}[\mathrm{R}]}{\mathrm{d} t}=k \times 1 \ & \mathrm{~d}[\mathrm{R}]=-k \mathrm{~d} t \end{aligned} $$
Integrating both sides
$$ \begin{equation*} [\mathrm{R}]=-k t+\mathrm{I} \tag{4.5} \end{equation*} $$
where, I is the constant of integration.
At $t=0$, the concentration of the reactant $R=R_0$, where $R_0$ is initial concentration of the reactant.
Substituting in equation (4.5)
$$ \begin{aligned} & {[\mathrm{R}]_0=-k \times 0+\mathrm{I}} \\ & {[\mathrm{R}]_0=\mathrm{I}} \end{aligned} $$
Substituting the value of I in the equation (4.5)
$$ \begin{equation*} \mathrm{R}=-k t+\mathrm{R}_0 \tag{4.6} \end{equation*} $$
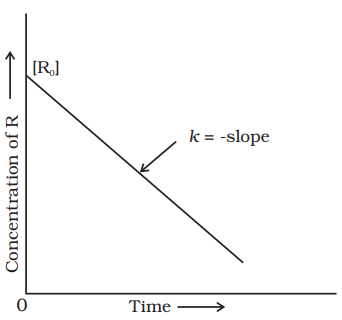
Comparing (4.6) with equation of a straight line, $y=m x+c$, if we plot $[R]$ against $t$, we get a straight line (Fig. 4.3) with slope $=-k$ and intercept equal to $[\mathrm{R}]_{0}$.
Further simplifying equation (4.6), we get the rate constant, $k$ as
$$ \begin{equation*} k=\frac{[\mathrm{R}]_{0}-[\mathrm{R}]}{t} \tag{4.7} \end{equation*} $$
Zero order reactions are relatively uncommon but they occur under special conditions. Some enzyme catalysed reactions and reactions which occur on metal surfaces are a few examples of zero order reactions. The decomposition of gaseous ammonia on a hot platinum surface is a zero order reaction at high pressure.
$$ 2 \mathrm{NH_3}(\mathrm{~g}) \underset{\text { Pt catalyst }}{1130 \mathrm{~K}} \rightarrow \mathrm{N_2}(\mathrm{~g})+3 \mathrm{H_2}(\mathrm{~g}) $$
$$ \text { Rate }=k\left[\mathrm{NH_3}\right]^{0}=k $$
In this reaction, platinum metal acts as a catalyst. At high pressure, the metal surface gets saturated with gas molecules. So, a further change in reaction conditions is unable to alter the amount of ammonia on the surface of the catalyst making rate of the reaction independent of its concentration. The thermal decomposition of HI on gold surface is another example of zero order reaction.
4.3.2 First Order Reactions
In this class of reactions, the rate of the reaction is proportional to the first power of the concentration of the reactant R. For example,
$$ \begin{aligned} & \mathrm{R} \rightarrow \mathrm{P} \\ & \text { Rate }=-\frac{\mathrm{d}[\mathrm{R}]}{\mathrm{d} t}=k[\mathrm{R}] \\ & \text { or } \frac{\mathrm{d}[\mathrm{R}]}{[\mathrm{R}]}=-k \mathrm{~d} t \end{aligned} $$
Integrating this equation, we get $$ \begin{equation*} \ln [R]=-k t+I \tag{4.8} \end{equation*} $$ Again, I is the constant of integration and its value can be determined easily.
When $t=0, R=[R]_0$, where $[R]_0$ is the initial concentration of the reactant.
Therefore, equation (3.8) can be written as
$$ \begin{aligned} & \ln [\mathrm{R}]_0=-k \times 0+\mathrm{I} \\ & \ln [\mathrm{R}]_0=\mathrm{I} \end{aligned} $$
Substituting the value of I in equation (3.8)
$$ \begin{equation*} \ln [R]=-k t+\ln [R]_0 \tag{4.9} \end{equation*} $$
Rearranging this equation
$$ \begin{align*} & \ln \frac{[\mathrm{R}]}{[\mathrm{R}]_0}=-k t \\ & \text { or } k=\frac{1}{t} \ln \frac{[\mathrm{R}]_0}{[\mathrm{R}]} \tag{4.10} \end{align*} $$
At time $t_1$ from equation (3.8)
$* \ln [\mathrm{R}]_1=-k t_1+{ }^* \ln [\mathrm{R}]_0$
At time $t_2$
$$ \begin{equation*} \ln [R]_2=-k t_2+\ln [R]_0 \tag{4.12} \end{equation*} $$
where, $[R]_1$ and $[R]_2$ are the concentrations of the reactants at time $t_1$ and $t_2$ respectively.
Subtracting (4.12) from (4.11)
$$ \begin{align*} & \ln [\mathrm{R}]_1-\ln [\mathrm{R}]_2=-k t_1-\left(-k t_2\right) \end{align*} $$
$$ \begin{align*} & \ln \frac{[\mathrm{R}]_1}{[\mathrm{R}]_2}=k\left(t_2-t_1\right) \\ & k=\frac{1}{\left(t_2-t_1\right)} \ln \frac{[\mathrm{R}]_1}{[\mathrm{R}]_2} \tag{4.13} \end{align*} $$
Equation (3.9) can also be written as
$$ \ln \frac{[\mathrm{R}]}{[\mathrm{R}]_{0}}=-k t $$
Taking antilog of both sides
$$ \begin{equation*} [\mathrm{R}]=[\mathrm{R}]_{0} \mathrm{e}^{-k t} \tag{4.14} \end{equation*} $$
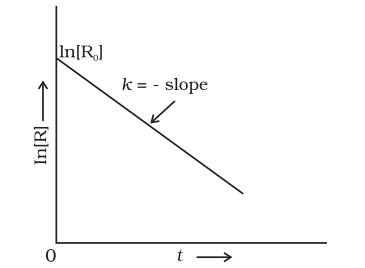
Comparing equation (3.9) with $\mathrm{y}=\mathrm{mx}+\mathrm{c}$, if we plot $\ln [R]$ against $t$ (Fig. 3.4) we get a straight line with slope $=-k$ and intercept equal to $\ln [\mathrm{R}]_{0}$
The first order rate equation (3.10) can also be written in the form
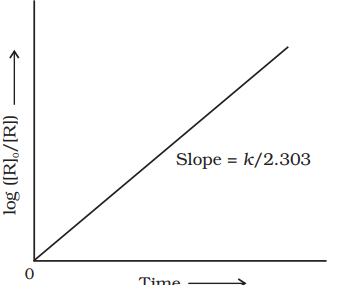
$$ \begin{align*} & k=\frac{2.303}{t} \log \frac{[\mathrm{R}]_0}{[\mathrm{R}]} \tag{4.15}\\ & * \log \frac{[\mathrm{R}]_0}{[\mathrm{R}]}=\frac{k t}{2.303} \end{align*} $$
If we plot a graph between $\log [R]_{0} /[R]$ vs $t$, (Fig. 3.5), the slope $=k / 2.303$
Hydrogenation of ethene is an example of first order reaction.
$$ \mathrm{C_2} \mathrm{H_4}(\mathrm{~g})+\mathrm{H_2}(\mathrm{~g}) \rightarrow \mathrm{C_2} \mathrm{H_6}(\mathrm{~g}) $$
Rate $=k\left[\mathrm{C_2} \mathrm{H_4}\right]$
All natural and artificial radioactive decay of unstable nuclei take place by first order kinetics.
Let us consider a typical first order gas phase reaction
$$ \mathrm{A}(\mathrm{g}) \rightarrow \mathrm{B}(\mathrm{g})+\mathrm{C}(\mathrm{g}) $$
Let $p_{\mathrm{i}}$ be the initial pressure of $\mathrm{A}$ and $p_{\mathrm{t}}$ the total pressure at time ’ $t$ ‘. Integrated rate equation for such a reaction can be derived as
Total pressure $p_{\mathrm{t}}=p_{\mathrm{A}}+p_{\mathrm{B}}+p_{\mathrm{C}}$ (pressure units) $p_{\mathrm{A}}, p_{\mathrm{B}}$ and $p_{\mathrm{C}}$ are the partial pressures of $\mathrm{A}, \mathrm{B}$ and $\mathrm{C}$, respectively.
If $\mathrm{x}$ atm be the decrease in pressure of $\mathrm{A}$ at time $t$ and one mole each of $\mathrm{B}$ and $\mathrm{C}$ is being formed, the increase in pressure of $\mathrm{B}$ and $\mathrm{C}$ will also be $\mathrm{x}$ atm each.
| $\mathrm{A}(\mathrm{g})$ | $\rightarrow$ | B(g) | + | $\mathrm{C}(\mathrm{g})$ | |
|---|---|---|---|---|---|
| At $t=0$ | $p_{\mathrm{i}}$ atm | $0 \mathrm{~atm}$ | $0 \mathrm{~atm}$ | ||
| At time | $\left(p_{\mathrm{i}}-\mathrm{x}\right) \mathrm{atm}$ | $\mathrm{x}$ atm | $\mathrm{x} \mathrm{atm}$ |
where, $p_{\mathrm{i}}$ is the initial pressure at time $t=0$.
$$ \begin{aligned} & p_{\mathrm{t}}=\left(p_{\mathrm{i}}-\mathrm{x}\right)+\mathrm{x}+\mathrm{x}=p_{\mathrm{i}}+\mathrm{x} \ & \mathrm{x}=\left(p_{\mathrm{t}}-p_{\mathrm{i}}\right) \end{aligned} $$
$$ \text { where, } \begin{align*} p_{\mathrm{A}} & =p_{\mathrm{i}}-\mathrm{x}=p_{\mathrm{i}}-\left(p_{\mathrm{t}}-p_{\mathrm{i}}\right) \\ & =2 p_{\mathrm{i}}-p_{\mathrm{t}} \\ k & =\left(\frac{2.303}{t}\right)\left(\log \frac{p_{\mathrm{i}}}{p_{\mathrm{A}}}\right) \tag{4.16}\\ = & \frac{2.303}{t} \log \frac{p_{\mathrm{i}}}{\left(2 p_{\mathrm{i}}-p_{\mathrm{t}}\right)} \end{align*} $$
The following data were obtained during the first order thermal decomposition of $\mathrm{N_2} \mathrm{O_5}(\mathrm{~g})$ at constant volume:
4.3.3 Half-Life of a Reaction
The half-life of a reaction is the time in which the concentration of a reactant is reduced to one half of its initial concentration. It is represented as t1/2.
$$ \begin{aligned} & k=\frac{[\mathrm{R}]_0-[\mathrm{R}]}{t} \\ \end{aligned} $$
$$ \begin{aligned} & \text { At } t=t_{1 / 2}, \quad[\mathrm{R}]=\frac{1}{2}[\mathrm{R}]_0 \end{aligned} $$
The rate constant at $t_{1 / 2}$ becomes
$$ \begin{aligned} k=\frac{[\mathrm{R}]_0- 1/ 2[\mathrm{R}]_0}{t_1/2} \end{aligned} $$
$$ \begin{aligned} & t_{1 / 2}=\frac{[\mathrm{R}]_0}{2 k} \end{aligned} $$
It is clear that $t_{1 / 2}$ for a zero order reaction is directly proportional to the initial concentration of the reactants and inversely proportional to the rate constant.
For the first order reaction,
$$ \begin{align*} & k=\frac{2.303}{t} \log \frac{[\mathrm{R}]_0}{[\mathrm{R}]} \tag{4.15}\\ & \text { at } \end{align*} $$
$$ \begin{align*} t_{1 / 2} \quad[\mathrm{R}]=\frac{[\mathrm{R}]_0}{2} \tag{4.16} \end{align*} $$
So, the above equation becomes
$$ \begin{align*} k & =\frac{2.303}{t_{1 / 2}} \log \frac{[\mathrm{R}]_{0}}{[\mathrm{R}] / 2} \\ \end{align*} $$
$$ \begin{align*} \text { or } t_{1 / 2} & =\frac{2.303}{k} \log 2 \\ \end{align*} $$
$$ \begin{align*} t_{1 / 2} & =\frac{2.303}{k} \times 0.301 \\ \end{align*} $$
$$ \begin{align*} t_{1 / 2} & =\frac{0.693}{k} \tag{4.17} \end{align*} $$
It can be seen that for a first order reaction, half-life period is constant, i.e., it is independent of initial concentration of the reacting species. The half-life of a first order equation is readily calculated from the rate constant and vice versa.
For zero order reaction $t_{1 / 2}\propto$ $R_0$. For first order reaction $t_{1 / 2}$ is independent of $[R]_0$.
The order of a reaction is sometimes altered by conditions. There are many reactions which obey first order rate law although they are higher order reactions. Consider the hydrolysis of ethyl acetate which is a chemical reaction between ethyl acetate and water. In reality, it is a second order reaction and concentration of both ethyl acetate and water affect the rate of the reaction. But water is taken in large excess for hydrolysis, therefore, concentration of water is not altered much during the reaction. Thus, the rate of reaction is affected by concentration of ethyl acetate only. For example, during the hydrolysis of $0.01 \mathrm{~mol}$ of ethyl acetate with $10 \mathrm{~mol}$ of water, amounts of the reactants and products at the beginning $(t=0)$ and completion $(t)$ of the reaction are given as under.
$$ \begin{array}{lllcc} & \mathrm{CH}_3 \mathrm{COOC}_2 \mathrm{H}_5 & +\mathrm{H}_2 \mathrm{O} \xrightarrow[\mathrm{H}^{+}]{ } \rightarrow & \mathrm{CH}_3 \mathrm{COOH} & +\mathrm{C}_2 \mathrm{H}_5 \mathrm{OH} \\ t=0 & 0.01 \mathrm{~mol} & 10 \mathrm{~mol} & 0 \mathrm{~mol} & 0 \mathrm{~mol} \\ t & 0 \mathrm{~mol} & 9.99 \mathrm{~mol} & 0.01 \mathrm{~mol} & 0.01 \mathrm{~mol} \end{array} $$
The concentration of water does not get altered much during the course of the reaction. So, the reaction behaves as first order reaction. Such reactions are called pseudo first orderreactions.
Inversion of cane sugar is another pseudo first order reaction.
$$ \begin{aligned} & C_{12} H_{22} O_{11}+H_2 {O} \xrightarrow{{H}^+} C_6 H_{12} O_6+C_6 H_{12} {O}_6 & \end{aligned} $$
$ \begin{aligned} \text { \quad \quad \quad \quad \quad \quad \quad \quad \quad \quad Cane sugar \quad \quad \quad \quad \quad Glucose \quad Fructose } \end{aligned} $
$$ \begin{aligned} \text { Rate }=k \quad\left[C_{12} H_{22} O_{11}\right] \end{aligned} $$
4.4 Temperature Dependence of the Rate of a Reaction
Most of the chemical reactions are accelerated by increase in temperature. For example, in decomposition of $\mathrm{N_2} \mathrm{O_5}$, the time taken for half of the original amount of material to decompose is $12 \mathrm{~min}$ at $50^{\circ} \mathrm{C}, 5 \mathrm{~h}$ at $25^{\circ} \mathrm{C}$ and 10 days at $0^{\circ} \mathrm{C}$. You also know that in a mixture of potassium permanganate $\left(\mathrm{KMnO_4}\right)$ and oxalic acid $\left(\mathrm{H_2} \mathrm{C_2} \mathrm{O_4}\right)$, potassium permanganate gets decolourised faster at a higher temperature than that at a lower temperature.
It has been found that for a chemical reaction with rise in temperature by $10^{\circ}$, the rate constant is nearly doubled.
The temperature dependence of the rate of a chemical reaction can be accurately explained by Arrhenius equation (4.18). It was first proposed by Dutch chemist, J.H. van’t Hoff but Swedish chemist, Arrhenius provided its physical justification and interpretation.
$$ \begin{equation*} k=\mathrm{A} \mathrm{e}^{-E \mathrm{a} / R T} \tag{4.18} \end{equation*} $$
where $A$ is the Arrhenius factor or the frequency factor. It is also called pre-exponential factor. It is a constant specific to a particular reaction. $R$ is gas constant and $E_{\mathrm{a}}$ is activation energy measured in joules/mole ( $\mathrm{J} \mathrm{mol}^{-1}$ ).
It can be understood clearly using the following simple reaction
$$ \mathrm{H_2}(\mathrm{~g})+\mathrm{I_2}(\mathrm{~g}) \rightarrow 2 \mathrm{HI}(\mathrm{g}) $$
According to Arrhenius, this reaction can take place only when a molecule of hydrogen and a molecule of iodine collide to form an unstable intermediate (Fig. 4.6). It exists for a very short time and then breaks up to form two molecules of hydrogen iodide.
The energy required to form this intermediate, called activated complex (C), is known as activation energy $\left(\boldsymbol{E_\mathrm{a}}\right).$ Fig. 4.7 is obtained by plotting potential energy vs reaction coordinate. Reaction coordinate represents the profile of energy change when reactants change into products.
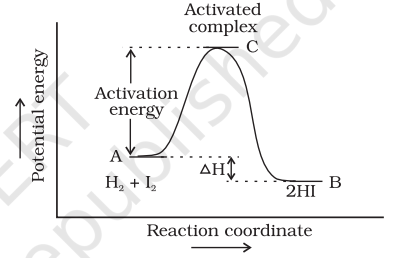
Some energy is released when the complex decomposes to form products. So, the final enthalpy of the reaction depends upon the nature of reactants and products.
All the molecules in the reacting species do not have the same kinetic energy. Since it is difficult to predict the behaviour of any one molecule with precision, Ludwig Boltzmann and James Clark Maxwell used statistics to predict the behaviour of large number of molecules. According to them, the distribution of kinetic energy may be described by plotting the fraction of molecules $\left(N_{\mathrm{E}} / N_{\mathrm{T}}\right)$ with a given kinetic energy (E) vs kinetic energy (Fig. 4.8). Here, $N_{\mathrm{E}}$ is the number of molecules with energy $E$ and $N_{\mathrm{T}}$ is total number of molecules.
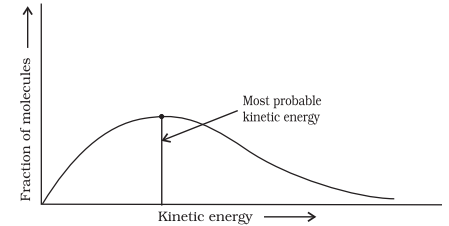
The peak of the curve corresponds to the most probable kinetic energy, i.e., kinetic energy of maximum fraction of molecules. There are decreasing number of molecules with energies higher or lower than this value. When the temperature is raised, the maximum of the curve moves to the higher energy value (Fig. 4.9) and the curve broadens out, i.e., spreads to the right such that there is a greater proportion of molecules with much higher energies. The area under the curve must be constant since total probability must be one at all times. We can mark the position of Ea on Maxwell Boltzmann distribution curve (Fig. 4.9).
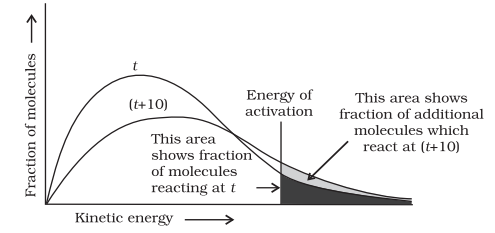
Increasing the temperature of the substance increases the fraction of molecules, which collide with energies greater than Ea. It is clear from the diagram that in the curve at (t + 10), the area showing the fraction of molecules having energy equal to or greater than activation energy gets doubled leading to doubling the rate of a reaction.
In the Arrhenius equation (4.18) the factor $\mathrm{e}^{-E \mathrm{Ea} / R T}$ corresponds to the fraction of molecules that have kinetic energy greater than $E_{\mathrm{a}}$. Taking natural logarithm of both sides of equation (4.18)
$$ \begin{equation*} \ln k=-\frac{E_{\mathrm{a}}}{R T}+\ln A \tag{4.19} \end{equation*} $$
The plot of $\ln k \mathrm{vs} 1 / \mathrm{T}$ gives a straight line according to the equation (3.19) as shown in Fig. 4.10.
Thus, it has been found from Arrhenius equation (4.18) that increasing the temperature or decreasing the activation energy will result in an increase in the rate of the reaction and an exponential increase in the rate constant.
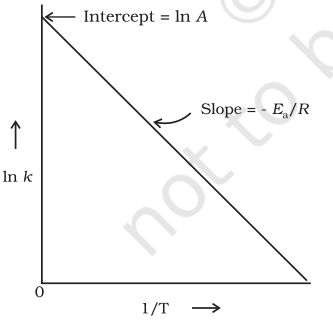
In Fig. 4.10, slope $=-\frac{E_{\mathrm{a}}}{R}$ and intercept $=\ln$
$A$. So we can calculate $E_{\mathrm{a}}$ and $A$ using these values.
At temperature $T_{1}$, equation (4.19) is
$$ \begin{equation*} \ln k_{1}=-\frac{E_{\mathrm{a}}}{R T_{1}}+\ln A \tag{4.20} \end{equation*} $$
At temperature $T_{2}$, equation (4.19) is
$$ \begin{equation*} \ln k_{2}=-\frac{E_{\mathrm{a}}}{R T_{2}}+\ln A \tag{4.21} \end{equation*} $$
(since $A$ is constant for a given reaction)
$k_{1}$ and $k_{2}$ are the values of rate constants at temperatures $T_{1}$ and $T_{2}$ respectively.
Subtracting equation (4.20) from (4.21), we obtain
$\ln k_{2}-\ln k_{1}=\frac{E_{\mathrm{a}}}{R T_{1}}-\frac{E_{\mathrm{a}}}{R T_{2}}$
$\ln \frac{k_{2}}{k_{1}}=\frac{E_{\mathrm{a}}}{R} \frac{1}{T_{1}}-\frac{1}{T_{2}}$
$\log \frac{k_{2}}{k_{1}}=\frac{E_{\mathrm{a}}}{2.303 R} \frac{1}{T_{1}}-\frac{1}{T_{2}}$
$\log \frac{k_{2}}{k_{1}}=\frac{E_{\mathrm{a}}}{2.303 \mathrm{R}} \frac{T_{2}-T_{1}}{T_{1} T_{2}}$
4.4.1 Effect of Catalyst
A catalyst is a substance which increases the rate of a reaction without itself undergoing any permanent chemical change. For example, $\mathrm{MnO_2}$ catalyses the following reaction so as to increase its rate considerably.
$$ 2 \mathrm{KClO_3} \xrightarrow{\mathrm{MnO_2}} 2 \mathrm{KCl}+3 \mathrm{O_2} $$
The word catalyst should not be used when the added substance reduces the rate of raction. The substance is then called inhibitor. The action of the catalyst can be explained by intermediate complex theory. According to this theory, a catalyst participates in a chemical reaction by forming temporary bonds with the reactants resulting in an intermediate complex. This has a transitory existence and decomposes to yield products and the catalyst.
It is believed that the catalyst provides an alternate pathway or reaction mechanism by reducing the activation energy between reactants and products and hence lowering the potential energy barrier as shown in Fig. 4.11.
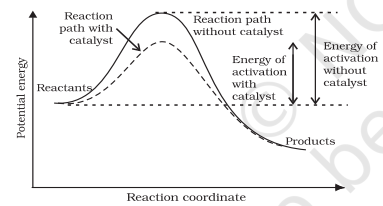
It is clear from Arrhenius equation (4.18) that lower the value of activation energy faster will be the rate of a reaction.
A small amount of the catalyst can catalyse a large amount of reactants. A catalyst does not alter Gibbs energy, DG of a reaction. It catalyses the spontaneous reactions but does not catalyse non-spontaneous reactions. It is also found that a catalyst does not change the equilibrium constant of a reaction rather, it helps in attaining the equilibrium faster, that is, it catalyses the forward as well as the backward reactions to the same extent so that the equilibrium state remains same but is reached earlier.
4.5 Collision Theory of Chemical Reactions
Though Arrhenius equation is applicable under a wide range of circumstances, collision theory, which was developed by Max Trautz and William Lewis in 1916 -18, provides a greater insight into the energetic and mechanistic aspects of reactions. It is based on kinetic theory of gases. According to this theory, the reactant molecules are assumed to be hard spheres and reaction is postulated to occur when molecules collide with each other. The number of collisions per second per unit volume of the reaction mixture is known as collision frequency (Z). Another factor which affects the rate of chemical reactions is activation energy (as we have already studied). For a bimolecular elementary reaction
$$ \mathrm{A}+\mathrm{B} \rightarrow \text { Products } $$
rate of reaction can be expressed as
$$ \begin{equation*} \text { Rate }=Z_{\mathrm{AB}} \mathrm{e}^{-E_{\mathrm{a}} / R T} \tag{4.23} \end{equation*} $$
where $Z_{\mathrm{AB}}$ represents the collision frequency of reactants, $\mathrm{A}$ and $\mathrm{B}$ and $\mathrm{e}^{-E a / R T}$ represents the fraction of molecules with energies equal to or greater than $E_{\mathrm{a}}$. Comparing (4.23) with Arrhenius equation, we can say that $A$ is related to collision frequency.
Equation (4.23) predicts the value of rate constants fairly accurately for the reactions that involve atomic species or simple molecules but for complex molecules significant deviations are observed. The reason could be that all collisions do not lead to the formation of products. The collisions in which molecules collide with sufficient kinetic energy (called threshold energy*) and proper orientation, so as to facilitate breaking of bonds between reacting species and formation of new bonds to form products are called as effective collisions.
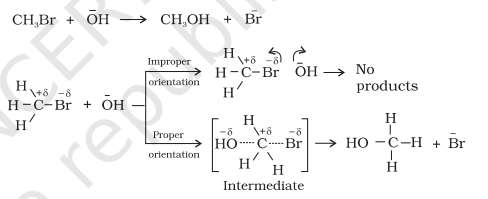
For example, formation of $\mathrm{CH_3} \mathrm{Br}+\overline{\mathrm{O}} \mathrm{H} \longrightarrow \mathrm{CH_3} \mathrm{OH}+\overline{\mathrm{Br}}$ methanol from bromoethane depends upon the orientation of reactant molecules as shown in Fig. 3.12. The proper orientation of reactant molecules lead to bond formation whereas improper orientation makes them simply bounce back and no products are formed.
To account for effective collisions, another factor P, called the probability or steric factor is introduced. It takes into account the fact that in a collision, molecules must be properly oriented i.e.,
$$ \text { Rate }=P Z_{\mathrm{AB}} \mathrm{e}^{-E_{\mathrm{a}} / R T} $$
Thus, in collision theory activation energy and proper orientation of the molecules together determine the criteria for an effective collision and hence the rate of a chemical reaction.
Collision theory also has certain drawbacks as it considers atoms/ molecules to be hard spheres and ignores their structural aspect. You will study details about this theory and more on other theories in your higher classes.
Summary
Chemical kinetics is the study of chemical reactions with respect to reaction rates, effect of various variables, rearrangement of atoms and formation of intermediates. The rate of a reaction is concerned with decrease in concentration of reactants or increase in the concentration of products per unit time. It can be expressed as instantaneous rate at a particular instant of time and average rate over a large interval of time. A number of factors such as temperature, concentration of reactants, catalyst, affect the rate of a reaction. Mathematical representation of rate of a reaction is given by rate law. It has to be determined experimentally and cannot be predicted. Order of a reaction with respect to a reactant is the power of its concentration which appears in the rate law equation. The order of a reaction is the sum of all such powers of concentration of terms for different reactants. Rate constant is the proportionality factor in the rate law. Rate constant and order of a reaction can be determined from rate law or its integrated rate equation. Molecularity is defined only for an elementary reaction. Its values are limited from 1 to 3 whereas order can be 0, 1, 2, 3 or even a fraction. Molecularity and order of an elementary reaction are same.
Temperature dependence of rate constants is described by Arrhenius equation $\left(k=A \mathrm{e}^{-E \mathrm{a} / R T}\right) . \quad E_{\mathrm{a}}$ corresponds to the activation energy and is given by the energy difference between activated complex and the reactant molecules, and $A$ (Arrhenius factor or pre-exponential factor) corresponds to the collision frequency. The equation clearly shows that increase of temperature or lowering of $\mathrm{E_a}$ will lead to an increase in the rate of reaction and presence of a catalyst lowers the activation energy by providing an alternate path for the reaction. According to collision theory, another factor $P$ called steric factor which refers to the orientation of molecules which collide, is important and contributes to effective collisions, thus, modifying the Arrhenius equation to $k=P Z_{\mathrm{AB}} \mathrm{e}^{-E_{\mathrm{a}} / R T}$.





