States of Matter(Deleted)
“The snowflake falls, yet lays not long Its feath’ry grasp on Mother Earth Ere Sun returns it to the vapors Whence it came, Or to waters tumbling down the rocky slope.”
Rod O’ Connor
INTRODUCTION
In previous units we have learnt about the properties related to single particle of matter, such as atomic size, ionization enthalpy, electronic charge density, molecular shape and polarity, etc. Most of the observable characteristics of chemical systems with which we are familiar represent bulk properties of matter, i.e., the properties associated with a collection of a large number of atoms, ions or molecules. For example, an individual molecule of a liquid does not boil but the bulk boils. Collection of water molecules have wetting properties; individual molecules do not wet. Water can exist as ice, which is a solid; it can exist as liquid; or it can exist in the gaseous state as water vapour or steam. Physical properties of ice, water and steam are very different. In all the three states of water chemical composition of water remains the same i.e.,
Chemical properties of a substance do not change with the change of its physical state; but rate of chemical reactions do depend upon the physical state. Many times in calculations while dealing with data of experiments we require knowledge of the state of matter. Therefore, it becomes necessary for a chemist to know the physical laws which govern the behaviour of matter in different states. In this unit, we will learn more about these three physical states of matter particularly liquid and gaseous states. To begin with, it is necessary to understand the nature of intermolecular forces, molecular interactions and effect of thermal energy on the motion of particles because a balance between these determines the state of a substance.
5.1 INTERMOLECULAR FORCES
Intermolecular forces are the forces of attraction and repulsion between interacting particles (atoms and molecules). This term does not include the electrostatic forces that exist between the two oppositely charged ions and the forces that hold atoms of a molecule together i.e., covalent bonds.
Attractive intermolecular forces are known as van der Waals forces, in honour of Dutch scientist Johannes van der Waals (18371923), who explained the deviation of real gases from the ideal behaviour through these forces. We will learn about this later in this unit. van der Waals forces vary considerably in magnitude and include dispersion forces or London forces, dipole-dipole forces, and dipole-induced dipole forces. A particularly strong type of dipole-dipole interaction is hydrogen bonding. Only a few elements can participate in hydrogen bond formation, therefore it is treated as a separate category. We have already learnt about this interaction in Unit 4.
At this point, it is important to note that attractive forces between an ion and a dipole are known as ion-dipole forces and these are not van der Waals forces. We will now learn about different types of van der Waals forces.
5.1.1 Dispersion Forces or London Forces
Atoms and nonpolar molecules are electrically symmetrical and have no dipole moment because their electronic charge cloud is symmetrically distributed. But a dipole may develop momentarily even in such atoms and molecules. This can be understood as follows. Suppose we have two atoms ’
so happen that momentarily electronic charge distribution in one of the atoms, say ’
The temporary dipoles of atom ’

dipoles is known as London force. Another name for this force is dispersion force. These forces are always attractive and interaction energy is inversely proportional to the sixth power of the distance between two interacting particles (i.e.,
5.1.2 Dipole - Dipole Forces
Dipole-dipole forces act between the molecules possessing permanent dipole. Ends of the dipoles possess “partial charges” and these charges are shown by Greek letter delta (

proportional to
5.1.3 Dipole–Induced Dipole Forces
This type of attractive forces operate between the polar molecules having permanent dipole and the molecules lacking permanent dipole. Permanent dipole of the polar molecule induces dipole on the electrically neutral molecule by deforming its electronic cloud (Fig. 5.3). Thus an induced dipole is developed in the other molecule. In this case also interaction energy is proportional to

In this case also cumulative effect of dispersion forces and dipole-induced dipole interactions exists.
5.1.4 Hydrogen bond
As already mentioned in section (5.1); this is special case of dipole-dipole interaction. We have already learnt about this in Unit 4. This
is found in the molecules in which highly polar
Intermolecular forces discussed so far are all attractive. Molecules also exert repulsive forces on one another. When two molecules are brought into close contact with each other, the repulsion between the electron clouds and that between the nuclei of two molecules comes into play. Magnitude of the repulsion rises very rapidly as the distance separating the molecules decreases. This is the reason that liquids and solids are hard to compress. In these states molecules are already in close contact; therefore they resist further compression; as that would result in the increase of repulsive interactions.
5.2 THERMAL ENERGY
Thermal energy is the energy of a body arising from motion of its atoms or molecules. It is directly proportional to the temperature of the substance. It is the measure of average kinetic energy of the particles of the matter and is thus responsible for movement of particles. This movement of particles is called thermal motion.
5.3 INTERMOLECULAR FORCES vs THERMAL INTERACTIONS
We have already learnt that intermolecular forces tend to keep the molecules together but thermal energy of the molecules tends to keep them apart. Three states of matter are the result of balance between intermolecular forces and the thermal energy of the molecules.
When molecular interactions are very weak, molecules do not cling together to make liquid or solid unless thermal energy is reduced by lowering the temperature. Gases do not liquify on compression only, although molecules come very close to each other and intermolecular forces operate to the maximum. However, when thermal energy of molecules is reduced by lowering the temperature; the gases can be very easily liquified. Predominance of thermal energy and the molecular interaction energy of a substance in three states is depicted as follows :
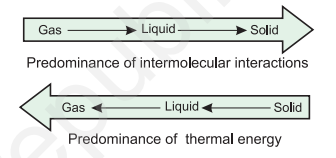
We have already learnt the cause for the existence of the three states of matter. Now we will learn more about gaseous and liquid states and the laws which govern the behaviour of matter in these states. We shall deal with the solid state in class XII.
5.4 THE GASEOUS STATE
This is the simplest state of matter. Throughout our life we remain immersed in the ocean of air which is a mixture of gases. We spend our life in the lowermost layer of the atmosphere called troposphere, which is held to the surface of the earth by gravitational force. The thin layer of atmosphere is vital to our life. It shields us from harmful radiations and contains substances like dioxygen, dinitrogen, carbon dioxide, water vapour, etc.
Let us now focus our attention on the behaviour of substances which exist in the gaseous state under normal conditions of temperature and pressure. A look at the periodic table shows that only eleven elements

exist as gases under normal conditions (Fig 5.4).
The gaseous state is characterized by the following physical properties.
- Gases are highly compressible.
- Gases exert pressure equally in all directions.
- Gases have much lower density than the solids and liquids.
- The volume and the shape of gases are not fixed. These assume volume and shape of the container.
- Gases mix evenly and completely in all proportions without any mechanical aid.
Simplicity of gases is due to the fact that the forces of interaction between their molecules are negligible. Their behaviour is governed by same general laws, which were discovered as a result of their experimental studies. These laws are relationships between measurable properties of gases. Some of these properties like pressure, volume, temperature and mass are very important because relationships between these variables describe state of the gas. Interdependence of these variables leads to the formulation of gas laws. In the next section we will learn about gas laws.
5.5 THE GAS LAWS
The gas laws which we will study now are the result of research carried on for several centuries on the physical properties of gases. The first reliable measurement on properties of gases was made by Anglo-Irish scientist Robert Boyle in 1662. The law which he formulated is known as Boyle’s Law. Later on attempts to fly in air with the help of hot air balloons motivated Jaccques Charles and Joseph Lewis Gay Lussac to discover additional gas laws. Contribution from Avogadro and others provided lot of information about gaseous state.
5.5.1 Boyle’s Law (Pressure - Volume Relationship)
On the basis of his experiments, Robert Boyle reached to the conclusion that at constant temperature, the pressure of a fixed amount (i.e., number of moles
where
On rearranging equation (5.2) we obtain
It means that at constant temperature, product of pressure and volume of a fixed amount of gas is constant.
If a fixed amount of gas at constant temperature

Figure 5.5 shows two conventional ways of graphically presenting Boyle’s law. Fig. 5.5 (a) is the graph of equation (5.3) at different temperatures. The value of
Fig 5.5 (b) represents the graph between
Experiments of Boyle, in a quantitative manner prove that gases are highly compressible because when a given mass of a gas is compressed, the same number of molecules occupy a smaller space. This means that gases become denser at high pressure. A relationship can be obtained between density and pressure of a gas by using Boyle’s law :
By definition, density ’
Table 5.1 Effect of Pressure on the Volume of
| Pressure |
Volume |
||
|---|---|---|---|
| 2.0 | 112.0 | 8.90 | 22.40 |
| 2.5 | 89.2 | 11.2 | 22.30 |
| 3.5 | 64.2 | 15.6 | 22.47 |
| 4.0 | 56.3 | 17.7 | 22.50 |
| 6.0 | 37.4 | 26.7 | 22.44 |
| 8.0 | 28.1 | 35.6 | 22.48 |
| 10.0 | 22.4 | 44.6 | 22.40 |
from Boyle’s law equation, we obtain the relationship.
This shows that at a constant temperature, pressure is directly proportional to the density of a fixed mass of the gas.
5.5.2 Charles’ Law (Temperature - Volume Relationship)
Charles and Gay Lussac performed several experiments on gases independently to improve upon hot air balloon technology. Their investigations showed that for a fixed mass of a gas at constant pressure, volume of a gas increases on increasing temperature and decreases on cooling. They found that for each degree rise in temperature, volume of a gas increases by
At this stage, we define a new scale of temperature such that
Thus
Thus we add 273 (more precisely 273.15) to the celsius temperature to obtain temperature at Kelvin scale.
If we write
in the equation (5.6) we obtain the relationship
Thus we can write a general equation as follows.
The value of constant
Equation (5.10) is the mathematical expression for Charles’ law, which states that pressure remaining constant, the volume
of a fixed mass of a gas is directly proportional to its absolute temperature. Charles found that for all gases, at any given pressure, graph of volume

Each line of the volume
Observations of Charles can be interpreted if we put the value of
All gases obey Charles’ law at very low pressures and high temperatures.
5.5.3 Gay Lussac’s Law (Pressure- Temperature Relationship)
Pressure in well inflated tyres of automobiles is almost constant, but on a hot summer day this increases considerably and tyre may burst if pressure is not adjusted properly. During winters, on a cold morning one may find the pressure in the tyres of a vehicle decreased considerably. The mathematical relationship between pressure and temperature was given by Joseph Gay Lussac and is known as Gay Lussac’s law. It states that at constant volume, pressure of a fixed amount of a gas varies directly with the temperature. Mathematically,
This relationship can be derived from Boyle’s law and Charles’ Law. Pressure vs temperature (Kelvin) graph at constant molar volume is shown in Fig. 5.7. Each line of this graph is called isochore.

5.5.4 Avogadro Law (Volume - Amount Relationship)
In 1811 Italian scientist Amedeo Avogadro tried to combine conclusions of Dalton’s atomic theory and Gay Lussac’s law of combining volumes (Unit 1) which is now known as Avogadro law. It states that equal volumes of all gases under the same conditions of temperature and pressure contain equal number of molecules. This means that as long as the temperature and pressure remain constant, the volume depends upon number of molecules of the gas or in other words amount of the gas. Mathematically we can write
The number of molecules in one mole of a gas has been determined to be
Since volume of a gas is directly proportional to the number of moles; one mole of each gas at standard temperature and pressure (STP)[^0] will have same volume. Standard temperature and pressure means
Molar volume of some gases is given in (Table 5.2).
Table 5.2 Molar volume in litres per mole of some gases at
| Argon | 22.37 |
|---|---|
| Carbon dioxide | 22.54 |
| Dinitrogen | 22.69 |
| Dioxygen | 22.69 |
| Dihydrogen | 22.72 |
| Ideal gas | 22.71 |
Number of moles of a gas can be calculated as follows
Where
Thus,
Equation (5.13) can be rearranged as follows :
Here ’
A gas that follows Boyle’s law, Charles’ law and Avogadro law strictly is called an ideal gas. Such a gas is hypothetical. It is assumed that intermolecular forces are not present between the molecules of an ideal gas. Real gases follow these laws only under certain specific conditions when forces of interaction are practically negligible. In all other situations these deviate from ideal behaviour. You will learn about the deviations later in this unit.
5.6 IDEAL GAS EQUATION
The three laws which we have learnt till now can be combined together in a single equation which is known as ideal gas equation.
At constant
where
Equation (5.18) shows that the value of
At STP conditions used earlier (
Ideal gas equation is a relation between four variables and it describes the state of any gas, therefore, it is also called equation of state.
Let us now go back to the ideal gas equation. This is the relationship for the simultaneous variation of the variables. If temperature, volume and pressure of a fixed amount of gas vary from
Equation (5.19) is a very useful equation. If out of six, values of five variables are known, the value of unknown variable can be calculated from the equation (5.19). This equation is also known as Combined gas law.
5.6.1 Density and Molar Mass of a Gaseous Substance
Ideal gas equation can be rearranged as follows:
Replacing
On rearranging equation (5.21) we get the relationship for calculating molar mass of a gas.
5.6.2 Dalton’s Law of Partial Pressures
The law was formulated by John Dalton in 1801. It states that the total pressure exerted by the mixture of non-reactive gases is equal to the sum of the partial pressures of individual gases i.e., the pressures which these gases would exert if they were enclosed separately in the same volume and under the same conditions of temperature. In a mixture of gases, the pressure exerted by the individual gas is called partial pressure. Mathematically,
where
Gases are generally collected over water and therefore are moist. Pressure of dry gas can be calculated by subtracting vapour pressure of water from the total pressure of the moist gas which contains water vapours also. Pressure exerted by saturated water vapour is called aqueous tension. Aqueous tension of water at different temperatures is given in Table 5.3.
Table 5.3 Aqueous Tension of Water (Vapour Pressure) as a Function of Temperature
| Temp./K | Pressure/bar | Temp./K | Pressure/bar |
|---|---|---|---|
| 273.15 | 0.0060 | 295.15 | 0.0260 |
| 283.15 | 0.0121 | 297.15 | 0.0295 |
| 288.15 | 0.0168 | 299.15 | 0.0331 |
| 291.15 | 0.0204 | 301.15 | 0.0372 |
| 293.15 | 0.0230 | 303.15 | 0.0418 |
Partial pressure in terms of mole fraction Suppose at the temperature T, three gases, enclosed in the volume
where
On dividing
where
Thus,
Similarly for other two gases we can write
Thus a general equation can be written as
where
5.7 KINETIC MOLECULAR THEORY OF GASES
So far we have learnt the laws (e.g., Boyle’s law, Charles’ law etc.) which are concise statements of experimental facts observed in the laboratory by the scientists. Conducting careful experiments is an important aspect of scientific method and it tells us how the particular system is behaving under different conditions. However, once the experimental facts are established, a scientist is curious to know why the system is behaving in that way. For example, gas laws help us to predict that pressure increases when we compress gases but we would like to know what happens at molecular level when a gas is compressed? A theory is constructed to answer such questions. A theory is a model (i.e., a mental picture) that enables us to better understand our observations. The theory that attempts to elucidate the behaviour of gases is known as kinetic molecular theory.
Assumptions or postulates of the kineticmolecular theory of gases are given below. These postulates are related to atoms and molecules which cannot be seen, hence it is said to provide a microscopic model of gases.
-
Gases consist of large number of identical particles (atoms or molecules) that are so small and so far apart on the average that the actual volume of the molecules is negligible in comparison to the empty space between them. They are considered as point masses. This assumption explains the great compressibility of gases.
-
There is no force of attraction between the particles of a gas at ordinary temperature and pressure. The support for this assumption comes from the fact that gases expand and occupy all the space available to them.
-
Particles of a gas are always in constant and random motion. If the particles were at rest and occupied fixed positions, then a gas would have had a fixed shape which is not observed.
-
Particles of a gas move in all possible directions in straight lines. During their random motion, they collide with each other and with the walls of the container. Pressure is exerted by the gas as a result of collision of the particles with the walls of the container.
-
Collisions of gas molecules are perfectly elastic. This means that total energy of molecules before and after the collision remains same. There may be exchange of energy between colliding molecules, their individual energies may change, but the sum of their energies remains constant. If there were loss of kinetic energy, the motion of molecules will stop and gases will settle down. This is contrary to what is actually observed.
-
At any particular time, different particles in the gas have different speeds and hence different kinetic energies. This assumption is reasonable because as the particles collide, we expect their speed to change. Even if initial speed of all the particles was same, the molecular collisions will disrupt this uniformity. Consequently the particles must have different speeds, which go on changing constantly. It is possible to show that though the individual speeds are changing, the distribution of speeds remains constant at a particular temperature.
-
If a molecule has variable speed, then it must have a variable kinetic energy. Under these circumstances, we can talk only about average kinetic energy. In kinetic theory it is assumed that average kinetic energy of the gas molecules is directly proportional to the absolute temperature. It is seen that on heating a gas at constant volume, the pressure increases. On heating the gas, kinetic energy of the particles increases and these strike the walls of the container more frequently thus exerting more pressure.
Kinetic theory of gases allows us to derive theoretically, all the gas laws studied in the previous sections. Calculations and predictions based on kinetic theory of gases agree very well with the experimental observations and thus establish the correctness of this model.
5.8 BEHAVIOUR OF REAL GASES: DEVIATION FROM IDEAL GAS BEHAVIOUR
Our theoritical model of gases corresponds very well with the experimental observations. Difficulty arises when we try to test how far the relation

It can be seen easily that at constant temperature
Deviation from ideal behaviour also becomes apparent when pressure

It is found that real gases do not follow, Boyle’s law, Charles law and Avogadro law perfectly under all conditions. Now two questions arise.
(i) Why do gases deviate from the ideal behaviour?
(ii) What are the conditions under which gases deviate from ideality?
We get the answer of the first question if we look into postulates of kinetic theory once again. We find that two assumptions of the kinetic theory do not hold good. These are
(a) There is no force of attraction between the molecules of a gas.
(b) Volume of the molecules of a gas is negligibly small in comparison to the space occupied by the gas.
If assumption (a) is correct, the gas will never liquify. However, we know that gases do liquify when cooled and compressed. Also, liquids formed are very difficult to compress.
This means that forces of repulsion are powerful enough and prevent squashing of molecules in tiny volume. If assumption (b) is correct, the pressure vs volume graph of experimental data (real gas) and that theoritically calculated from Boyles law (ideal gas) should coincide.
Real gases show deviations from ideal gas law because molecules interact with each other. At high pressures molecules of gases are very close to each other. Molecular interactions start operating. At high pressure, molecules do not strike the walls of the container with full impact because these are dragged back by other molecules due to molecular attractive forces. This affects the pressure exerted by the molecules on the walls of the container. Thus, the pressure exerted by the gas is lower than the pressure exerted by the ideal gas.
Here, a is a constant.
Repulsive forces also become significant. Repulsive interactions are short-range interactions and are significant when molecules are almost in contact. This is the situation at high pressure. The repulsive forces cause the molecules to behave as small but impenetrable spheres. The volume occupied by the molecules also becomes significant because instead of moving in volume
Equation (5.31) is known as van der Waals equation. In this equation
Also, at very low temperature, intermolecular forces become significant. As the molecules travel with low average speed, these can be captured by one another due to attractive forces. Real gases show ideal behaviour when conditions of temperature and pressure are such that the intermolecular forces are practically negligible. The real gases show ideal behaviour when pressure approaches zero.
The deviation from ideal behaviour can be measured in terms of compressibility factor
For ideal gas

have
to it. In other words, the behaviour of the gas becomes more ideal when pressure is very low. Upto what pressure a gas will follow the ideal gas law, depends upon nature of the gas and its temperature. The temperature at which a real gas obeys ideal gas law over an appreciable range of pressure is called Boyle temperature or Boyle point. Boyle point of a gas depends upon its nature. Above their Boyle point, real gases show positive deviations from ideality and
More insight is obtained in the significance of
If the gas shows ideal behaviour then
From equation (5.34) we can see that compressibility factor is the ratio of actual molar volume of a gas to the molar volume of it, if it were an ideal gas at that temperature and pressure.
In the following sections we will see that it is not possible to distinguish between gaseous state and liquid state and that liquids may be considered as continuation of gas phase into a region of small volumes and very high molecular attraction. We will also see how we can use isotherms of gases for predicting the conditions for liquifaction of gases.
5.9 LIQUEFACTION OF GASES
First complete data on pressure - volume temperature relations of a substance in both gaseous and liquid state was obtained by Thomas Andrews on carbon dioxide. He plotted isotherms of carbon dioxide at various temperatures (Fig. 5.11). Later on it was found that real gases behave in the same manner as carbon dioxide. Andrews noticed that at high temperatures isotherms look like that of an ideal gas and the gas cannot be liquified even at very high pressure. As the temperature is lowered, shape of the curve changes and data shows considerable deviation from ideal behaviour. At

carbon dioxide remains gas upto 73 atmospheric pressure. (Point E in Fig. 5.11). At 73 atmospheric pressure, liquid carbon dioxide appears for the first time. The temperature
It is possible to change a gas into liquid or a liquid into gas by a process in which always a single phase is present. For example in Fig. 5.11 we can move from point
Thus there is continuity between the gaseous and liquid state. The term fluid is used for either a liquid or a gas to recognise this continuity. Thus a liquid can be viewed as a very dense gas. Liquid and gas can be distinguished only when the fluid is below its critical temperature and its pressure and volume lie under the dome, since in that situation liquid and gas are in equilibrium and a surface separating the two phases is visible. In the absence of this surface there is no fundamental way of distinguishing between two states. At critical temperature, liquid passes into gaseous state imperceptibly and continuously; the surface separating two phases disappears (Section 5.10.1). A gas below the critical temperature can be liquified by applying pressure, and is called vapour of the substance. Carbon dioxide gas below its critical temperature is called carbon dioxide vapour. Critical constants for some common substances are given in Table 5.4.
Table 5.4 Critical Constants for Some Substances
| Substance | |||
|---|---|---|---|
| 33.2 | 12.97 | 0.0650 | |
| 5.3 | 2.29 | 0.0577 | |
| 126. | 33.9 | 0.0900 | |
| 154.3 | 50.4 | 0.0744 | |
| 304.10 | 73.9 | 0.0956 | |
| 647.1 | 220.6 | 0.0450 | |
| 405.5 | 113.0 | 0.0723 |
5.10 LIGUID STATE
Intermolecular forces are stronger in liquid state than in gaseous state. Molecules in liquids are so close that there is very little empty space between them and under normal conditions liquids are denser than gases.
Molecules of liquids are held together by attractive intermolecular forces. Liquids have definite volume because molecules do not separate from each other. However, molecules of liquids can move past one another freely, therefore, liquids can flow, can be poured and can assume the shape of the container in which these are stored. In the following sections we will look into some of the physical properties of the liquids such as vapour pressure, surface tension and viscosity.
5.10.1 Vapour Pressure
If an evacuated container is partially filled with a liquid, a portion of liquid evaporates to fill the remaining volume of the container with vapour. Initially the liquid evaporates and pressure exerted by vapours on the walls of the container (vapour pressure) increases. After some time it becomes constant, an equilibrium is established between liquid phase and vapour phase. Vapour pressure at this stage is known as equilibrium vapour pressure or saturated vapour pressure.. Since process of vapourisation is temperature dependent; the temperature must be mentioned while reporting the vapour pressure of a liquid.
When a liquid is heated in an open vessel, the liquid vapourises from the surface. At the temperature at which vapour pressure of the liquid becomes equal to the external pressure, vapourisation can occur throughout the bulk of the liquid and vapours expand freely into the surroundings. The condition of free vapourisation throughout the liquid is called boiling. The temperature at which vapour pressure of liquid is equal to the external pressure is called boiling temperature at that pressure. Vapour pressure of some common liquids at various temperatures is given in (Fig. 5.12). At 1 atm pressure boiling temperature is called normal boiling point. If pressure is 1 bar then the boiling point is called standard boiling point of the liquid. Standard boiling point of the liquid is slightly lower than the normal boiling point because 1 bar pressure is slightly less than
At high altitudes atmospheric pressure is low. Therefore liquids at high altitudes boil at lower temperatures in comparison to that at sea level. Since water boils at low temperature on hills, the pressure cooker is used for cooking food. In hospitals surgical

instruments are sterilized in autoclaves in which boiling point of water is increased by increasing the pressure above the atmospheric pressure by using a weight covering the vent.
Boiling does not occur when liquid is heated in a closed vessel. On heating continuously vapour pressure increases. At first a clear boundary is visible between liquid and vapour phase because liquid is more dense than vapour. As the temperature increases more and more molecules go to vapour phase and density of vapours rises. At the same time liquid becomes less dense. It expands because molecules move apart. When density of liquid and vapours becomes the same; the clear boundary between liquid and vapours disappears. This temperature is called critical temperature about which we have already discussed in section 5.9.
5.10.2 Surface Tension
It is well known fact that liquids assume the shape of the container. Why is it then small drops of mercury form spherical bead instead of spreading on the surface. Why do particles of soil at the bottom of river remain separated but they stick together when taken out? Why does a liquid rise (or fall) in a thin capillary as soon as the capillary touches the surface of the liquid? All these phenomena are caused due to the characteristic property of liquids, called surface tension. A molecule in the bulk of liquid experiences equal intermolecular forces from all sides. The molecule, therefore does not experience any net force. But for the molecule on the surface of liquid, net attractive force is towards the interior of the liquid (Fig. 5.13), due to the molecules below it. Since there are no molecules above it.
Liquids tend to minimize their surface area. The molecules on the surface experience a net downward force and have more energy than the molecules in the bulk, which do not experience any net force. Therefore, liquids tend to have minimum number of molecules at their surface. If surface of the liquid is increased by pulling a molecule from the bulk, attractive forces will have to be overcome. This will require expenditure of energy. The energy required to increase the surface area of the liquid by one unit is defined as surface energy.

Its dimensions are
Liquid tends to rise (or fall) in the capillary because of surface tension. Liquids wet the things because they spread across their surfaces as thin film. Moist soil grains are pulled together because surface area of thin film of water is reduced. It is surface tension which gives stretching property to the surface of a liquid. On flat surface, droplets are slightly flattened by the effect of gravity; but in the gravity free environments drops are perfectly spherical.
The magnitude of surface tension of a liquid depends on the attractive forces between the molecules. When the attractive forces are large, the surface tension is large. Increase in temperature increases the kinetic energy of the molecules and effectiveness of intermolecular attraction decreases, so surface tension decreases as the temperature is raised.
5.10.3 Viscosity
It is one of the characteristic properties of liquids. Viscosity is a measure of resistance to flow which arises due to the internal friction between layers of fluid as they slip past one another while liquid flows. Strong intermolecular forces between molecules hold them together and resist movement of layers past one another.
When a liquid flows over a fixed surface, the layer of molecules in the immediate contact of surface is stationary. The velocity of upper layers increases as the distance of layers from the fixed layer increases. This type of flow in which there is a regular gradation of velocity in passing from one layer to the next is called laminar flow. If we choose any layer in the flowing liquid (Fig.5.14), the layer above it accelerates its flow and the layer below this retards its flow.

If the velocity of the layer at a distance
the change in velocity with distance)
’
1 poise
Greater the viscosity, the more slowly the liquid flows. Hydrogen bonding and van der Waals forces are strong enough to cause high viscosity. Glass is an extremely viscous liquid. It is so viscous that many of its properties resemble solids. However, property of flow of glass can be experienced by measuring the thickness of windowpanes of old buildings. These become thicker at the bottom than at the top.
Viscosity of liquids decreases as the temperature rises because at high temperature molecules have high kinetic energy and can overcome the intermolecular forces to slip past one another between the layers.
Summary
Intermolecular forces operate between the particles of matter. These forces differ from pure electrostatic forces that exist between two oppositely charged ions. Also, these do not include forces that hold atoms of a covalent molecule together through covalent bond. Competition between thermal energy and intermolecular interactions determines the state of matter. “Bulk” properties of matter such as behaviour of gases, characteristics of solids and liquids and change of state depend upon energy of constituent particles and the type of interaction between them. Chemical properties of a substance do not change with change of state, but the reactivity depends upon the physical state.
Forces of interaction between gas molecules are negligible and are almost independent of their chemical nature. Interdependence of some observable properties namely pressure, volume, temperature and mass leads to different gas laws obtained from experimental studies on gases. Boyle’s law states that under isothermal condition, pressure of a fixed amount of a gas is inversely proportional to its volume. Charles’ law is a relationship between volume and absolute temperature under isobaric condition. It states that volume of a fixed amount of gas is directly proportional to its absolute temperature
At high pressure and low temperature intermolecular forces start operating strongly between the molecules of gases because they come close to each other. Under suitable temperature and pressure conditions gases can be liquified. Liquids may be considered as continuation of gas phase into a region of small volume and very strong molecular attractions. Some properties of liquids e.g., surface tension and viscosity are due to strong intermolecular attractive forces.intermolecular attractive forces.





