Chemical Bonding and Moleuclar Structure
“Scientists are constantly discovering new compounds, orderly arranging the facts about them, trying to explain with the existing knowledge, organising to modify the earlier views or evolve theories for explaining the newly observed facts.”
Matter is made up of one or different type of elements. Under normal conditions no other element exists as an independent atom in nature, except noble gases. However, a group of atoms is found to exist together as one species having characteristic properties. Such a group of atoms is called a molecule. Obviously there must be some force which holds these constituent atoms together in the molecules. The attractive force which holds various constituents (atoms, ions, etc.) together in different chemical species is called a chemical bond. Since the formation of chemical compounds takes place as a result of combination of atoms of various elements in different ways, it raises many questions. Why do atoms combine? Why are only certain combinations possible? Why do some atoms combine while certain others do not? Why do molecules possess definite shapes? To answer such questions different theories and concepts have been put forward from time to time. These are Kössel-Lewis approach, Valence Shell Electron Pair Repulsion (VSEPR) Theory, Valence Bond (VB) Theory and Molecular Orbital (MO) Theory. The evolution of various theories of valence and the interpretation of the nature of chemical bonds have closely been related to the developments in the understanding of the structure of atom, the electronic configuration of elements and the periodic table. Every system tends to be more stable and bonding is nature’s way of lowering the energy of the system to attain stability.
4.1 KÖSSEL-LEWIS APPROACH TO CHEMICAL BONDING
In order to explain the formation of chemical bond in terms of electrons, a number of attempts were made, but it was only in 1916 when Kössel and Lewis succeeded independently in giving a satisfactory explanation. They were the first to provide some logical explanation of valence which was based on the inertness of noble gases.
Lewis pictured the atom in terms of a positively charged ‘Kernel’ (the nucleus plus the inner electrons) and the outer shell that could accommodate a maximum of eight electrons. He, further assumed that these eight electrons occupy the corners of a cube which surround the ‘Kernel’. Thus the single outer shell electron of sodium would occupy one corner of the cube, while in the case of a noble gas all the eight corners would be occupied. This octet of electrons, represents a particularly stable electronic arrangement. Lewis postulated that atoms achieve the stable octet when they are linked by chemical bonds. In the case of sodium and chlorine, this can happen by the transfer of an electron from sodium to chlorine thereby giving the
Lewis Symbols: In the formation of a molecule, only the outer shell electrons take part in chemical combination and they are known as valence electrons. The inner shell electrons are well protected and are generally not involved in the combination process. G.N. Lewis, an American chemist introduced simple notations to represent valence electrons in an atom. These notations are called Lewis symbols. For example, the Lewis symbols for the elements of second period are as under:

Significance of Lewis Symbols : The number of dots around the symbol represents the number of valence electrons. This number of valence electrons helps to calculate the common or group valence of the element. The group valence of the elements is generally either equal to the number of dots in Lewis symbols or 8 minus the number of dots or valence electrons.
Kössel, in relation to chemical bonding, drew attention to the following facts:
- In the periodic table, the highly electronegative halogens and the highly electropositive alkali metals are separated by the noble gases;
- The formation of a negative ion from a halogen atom and a positive ion from an alkali metal atom is associated with the gain and loss of an electron by the respective atoms;
- The negative and positive ions thus formed attain stable noble gas electronic configurations. The noble gases (with the exception of helium which has a duplet of electrons) have a particularly stable outer shell configuration of eight (octet) electrons,
- The negative and positive ions are stabilized by electrostatic attraction.
For example, the formation of
Similarly the formation of
The bond formed, as a result of the electrostatic attraction between the positive and negative ions was termed as the electrovalent bond. The electrovalence is thus equal to the number of unit charge(s) on the ion. Thus, calcium is assigned a positive electrovalence of two, while chlorine a negative electrovalence of one.
Kössel’s postulations provide the basis for the modern concepts regarding ion-formation by electron transfer and the formation of ionic crystalline compounds. His views have proved to be of great value in the understanding and systematisation of the ionic compounds. At the same time he did recognise the fact that a large number of compounds did not fit into these concepts.
4.1.1 Octet Rule
Kössel and Lewis in 1916 developed an important theory of chemical combination between atoms known as electronic theory of chemical bonding. According to this, atoms can combine either by transfer of valence electrons from one atom to another (gaining or losing) or by sharing of valence electrons in order to have an octet in their valence shells. This is known as octet rule.
4.1.2 Covalent Bond
Langmuir (1919) refined the Lewis postulations by abandoning the idea of the stationary cubical arrangement of the octet, and by introducing the term covalent bond. The Lewis-Langmuir theory can be understood by considering the formation of the chlorine molecule,
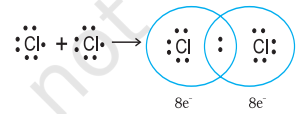
chlorine atoms attain the outer shell octet of the nearest noble gas (i.e., argon).
The dots represent electrons. Such structures are referred to as Lewis dot structures.
The Lewis dot structures can be written for other molecules also, in which the combining atoms may be identical or different. The important conditions being that:
- Each bond is formed as a result of sharing of an electron pair between the atoms.
- Each combining atom contributes at least one electron to the shared pair.
- The combining atoms attain the outershell noble gas configurations as a result of the sharing of electrons.
- Thus in water and carbon tetrachloride molecules, formation of covalent bonds can be represented as:

Thus, when two atoms share one electron pair they are said to be joined by a single covalent bond. In many compounds we have multiple bonds between atoms. The formation of multiple bonds envisages sharing of more than one electron pair between two atoms. If two atoms share two pairs of electrons, the covalent bond between them is called a double bond. For example, in the carbon dioxide molecule, we have two double bonds between the carbon and oxygen atoms. Similarly in ethene molecule the two carbon atoms are joined by a double bond..

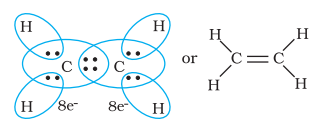
When combining atoms share three electron pairs as in the case of two nitrogen atoms in the
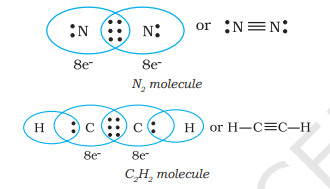
4.1.3 Lewis Representation of Simple Molecules (the Lewis Structures)
The Lewis dot structures provide a picture of bonding in molecules and ions in terms of the shared pairs of electrons and the octet rule. While such a picture may not explain the bonding and behaviour of a molecule completely, it does help in understanding the formation and properties of a molecule to a large extent. Writing of Lewis dot structures of molecules is, therefore, very useful. The Lewis dot structures can be written by adopting the following steps:
- The total number of electrons required for writing the structures are obtained by adding the valence electrons of the combining atoms. For example, in the
- For anions, each negative charge would mean addition of one electron. For cations, each positive charge would result in subtraction of one electron from the total number of valence electrons. For example, for the
- Knowing the chemical symbols of the combining atoms and having knowledge of the skeletal structure of the compound (known or guessed intelligently), it is easy to distribute the total number of electrons as bonding shared pairs between the atoms in proportion to the total bonds.
- In general the least electronegative atom occupies the central position in the molecule/ion. For example in the
- After accounting for the shared pairs of electrons for single bonds, the remaining electron pairs are either utilized for multiple bonding or remain as the lone pairs. The basic requirement being that each bonded atom gets an octet of electrons.
Lewis representations of a few molecules/ ions are given in Table 4.1.

4.1.4 Formal Charge
Lewis dot structures, in general, do not represent the actual shapes of the molecules. In case of polyatomic ions, the net charge is possessed by the ion as a whole and not by a particular atom. It is, however, feasible to assign a formal charge on each atom. The formal charge of an atom in a polyatomic molecule or ion may be defined as the difference between the number of valence electrons of that atom in an isolated or free state and the number of electrons assigned to that atom in the Lewis structure. It is expressed as :

The counting is based on the assumption that the atom in the molecule owns one electron of each shared pair and both the electrons of a lone pair.
Let us consider the ozone molecule
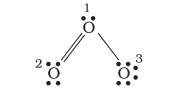
The atoms have been numbered as 1,2 and 3. The formal charge on:
- The central
- The end
Hence, we represent

We must understand that formal charges do not indicate real charge separation within the molecule. Indicating the charges on the atoms in the Lewis structure only helps in keeping track of the valence electrons in the molecule. Formal charges help in the selection of the lowest energy structure from a number of possible Lewis structures for a given species. Generally the lowest energy structure is the one with the smallest formal charges on the atoms. The formal charge is a factor based on a pure covalent view of bonding in which electron pairs are shared equally by neighbouring atoms.
4.1.5 Limitations of the Octet Rule
The octet rule, though useful, is not universal. It is quite useful for understanding the structures of most of the organic compounds and it applies mainly to the second period elements of the periodic table. There are three types of exceptions to the octet rule.
The incomplete octet of the central atom In some compounds, the number of electrons surrounding the central atom is less than eight. This is especially the case with elements having less than four valence electrons. Examples are

Odd-electron molecules In molecules with an odd number of electrons like nitric oxide,

The expanded octet
Elements in and beyond the third period of the periodic table have, apart from
Some of the examples of such compounds are:
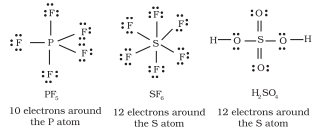
Interestingly, sulphur also forms many compounds in which the octet rule is obeyed. In sulphur dichloride, the
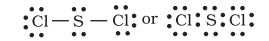
Other drawbacks of the octet theory
- It is clear that octet rule is based upon the chemical inertness of noble gases. However, some noble gases (for example xenon and krypton) also combine with oxygen and fluorine to form a number of compounds like
- This theory does not account for the shape of molecules.
- It does not explain the relative stability of the molecules being totally silent about the energy of a molecule.
4.2 IONIC OR ELECTROVALENT BOND
From the Kössel and Lewis treatment of the formation of an ionic bond, it follows that the formation of ionic compounds would primarily depend upon:
- The ease of formation of the positive and negative ions from the respective neutral atoms;
- The arrangement of the positive and negative ions in the solid, that is, the lattice of the crystalline compound.
The formation of a positive ion involves ionization, i.e., removal of electron(s) from the neutral atom and that of the negative ion involves the addition of electron(s) to the neutral atom.
| Ionization enthalpy | |||
| Electron gain enthalpy | |||
The electron gain enthalpy,
Obviously ionic bonds will be formed more easily between elements with comparatively low ionization enthalpies and elements with comparatively high negative value of electron gain enthalpy.
Most ionic compounds have cations derived from metallic elements and anions from non-metallic elements. The ammonium ion,
Ionic compounds in the crystalline state consist of orderly three-dimensional arrangements of cations and anions held together by coulombic interaction energies. These compounds crystallise in different crystal structures determined by the size of the ions, their packing arrangements and other factors. The crystal structure of sodium chloride,

In ionic solids, the sum of the electron gain enthalpy and the ionization enthalpy may be positive but still the crystal structure gets stabilized due to the energy released in the formation of the crystal lattice. For example: the ionization enthalpy for
Since lattice enthalpy plays a key role in the formation of ionic compounds, it is important that we learn more about it.
4.2.1 Lattice Enthalpy
The Lattice Enthalpy of an ionic solid is defined as the energy required to completely separate one mole of a solid ionic compound into gaseous constituent ions. For example, the lattice enthalpy of
This process involves both the attractive forces between ions of opposite charges and the repulsive forces between ions of like charge. The solid crystal being threedimensional; it is not possible to calculate lattice enthalpy directly from the interaction of forces of attraction and repulsion only. Factors associated with the crystal geometry have to be included.
4.3 BOND PARAMETERS
4.3.1 Bond Length
Bond length is defined as the equilibrium distance between the nuclei of two bonded atoms in a molecule. Bond lengths are measured by spectroscopic, X-ray diffraction and electron-diffraction techniques about which you will learn in higher classes. Each atom of the bonded pair contributes to the bond length (Fig. 4.1). In the case of a covalent bond, the contribution from each atom is called the covalent radius of that atom.
The covalent radius is measured approximately as the radius of an atom’s core which is in contact with the core of an adjacent atom in a bonded situation. The covalent radius is half of the distance between two similar atoms joined by a covalent bond

in the same molecule. The van der Waals radius represents the overall size of the atom which includes its valence shell in a nonbonded situation. Further, the van der Waals radius is half of the distance between two similar atoms in separate molecules in a solid. Covalent and van der Waals radii of chlorine are depicted in Fig. 4.2.

Some typical average bond lengths for single, double and triple bonds are shown in Table 4.2. Bond lengths for some common molecules are given in Table 4.3.
The covalent radii of some common elements are listed in Table 4.4.
4.3.2 Bond Angle
It is defined as the angle between the orbitals containing bonding electron pairs around the central atom in a molecule/complex ion. Bond angle is expressed in degree which can be experimentally determined by spectroscopic methods. It gives some idea regarding the distribution of orbitals around the central atom in a molecule/complex ion and hence it helps us in determining its shape. For example

4.3.3 Bond Enthalpy
It is defined as the amount of energy required to break one mole of bonds of a particular type between two atoms in a gaseous state. The unit of bond enthalpy is
Similarly the bond enthalpy for molecules containing multiple bonds, for example
It is important that larger the bond dissociation enthalpy, stronger will be the bond in the molecule. For a heteronuclear diatomic molecules like
In case of polyatomic molecules, the measurement of bond strength is more complicated. For example in case of
| Bond Type | Covalent Bond Length (pm) |
|---|---|
| 96 | |
| 107 | |
| 136 | |
| 143 | |
| 143 | |
| 154 | |
| 121 | |
| 122 | |
| 133 | |
| 138 | |
| 116 | |
| 120 |
Table 4.3 Bond Lengths in Some Common Molecules
| Molecule | Bond Length (pm) |
|---|---|
| 74 | |
| 144 | |
| 199 | |
| 228 | |
| 267 | |
| 109 | |
| 121 | |
| 92 | |
| 127 | |
| 141 | |
| 160 |
Table 4.4 Covalent Radii,
| 37 | |||||||
|---|---|---|---|---|---|---|---|
| 64 | |||||||
| 99 | |||||||
| 110 | 114 | ||||||
| 121 | 104 | 133 | |||||
| 141 | 137 |
The difference in the
4.3.4 Bond Order
In the Lewis description of covalent bond, the Bond Order is given by the number of bonds between the two atoms in a molecule. The bond order, for example in
Isoelectronic molecules and ions have identical bond orders; for example,
A general correlation useful for understanding the stablities of molecules is that: with increase in bond order, bond enthalpy increases and bond length decreases.
4.3.5 Resonance Structures
It is often observed that a single Lewis structure is inadequate for the representation of a molecule in conformity with its experimentally determined parameters. For example, the ozone,

In both structures we have a
The concept of resonance was introduced to deal with the type of difficulty experienced in the depiction of accurate structures of molecules like
Some of the other examples of resonance structures are provided by the carbonate ion and the carbon dioxide molecule.
In general, it may be stated that
- Resonance stabilizes the molecule as the energy of the resonance hybrid is less than the energy of any single cannonical structure; and,
- Resonance averages the bond characteristics as a whole.
Thus the energy of the
Many misconceptions are associated with resonance and the same need to be dispelled. You should remember that:
- The cannonical forms have no real existence.
- The molecule does not exist for a certain fraction of time in one cannonical form and for other fractions of time in other cannonical forms.
- There is no such equilibrium between the cannonical forms as we have between tautomeric forms (keto and enol) in tautomerism.
- The molecule as such has a single structure which is the resonance hybrid of the cannonical forms and which cannot as such be depicted by a single Lewis structure.
4.3.6 Polarity of Bonds
The existence of a hundred percent ionic or covalent bond represents an ideal situation. In reality no bond or a compound is either completely covalent or ionic. Even in case of covalent bond between two hydrogen atoms, there is some ionic character.
When covalent bond is formed between two similar atoms, for example in
As a result of polarisation, the molecule possesses the dipole moment (depicted below) which can be defined as the product of the magnitude of the charge and the distance between the centres of positive and negative charge. It is usually designated by a Greek letter ’
Dipole moment
Dipole moment is usually expressed in Debye units (D). The conversion factor is
where
Further dipole moment is a vector quantity and by convention it is depicted by a small arrow with tail on the negative centre and head pointing towards the positive centre. But in chemistry presence of dipole moment is represented by the crossed arrow
This arrow symbolises the direction of the shift of electron density in the molecule. Note that the direction of crossed arrow is opposite to the conventional direction of dipole moment vector
Peter Debye, the Dutch chemist received Nobel prize in 1936 for his work on X-ray diffraction and dipole moments. The magnitude of the dipole moment is given in Debye units in order to honour him.

In case of polyatomic molecules the dipole moment not only depend upon the individual dipole moments of bonds known as bond dipoles but also on the spatial arrangement of various bonds in the molecule. In such case, the dipole moment of a molecule is the vector sum of the dipole moments of various bonds. For example in
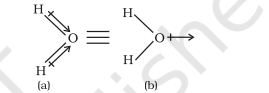
Net Dipole moment,
The dipole moment in case of

In tetra-atomic molecule, for example in

Let us study an interesting case of

Dipole moments of some molecules are shown in Table 4.5.
Just as all the covalent bonds have some partial ionic character, the ionic bonds also have partial covalent character. The partial covalent character of ionic bonds was discussed by Fajans in terms of the following rules:
- The smaller the size of the cation and the larger the size of the anion, the greater the covalent character of an ionic bond.
- The greater the charge on the cation, the greater the covalent character of the ionic bond.
- For cations of the same size and charge, the one, with electronic configuration
The cation polarises the anion, pulling the electronic charge toward itself and thereby increasing the electronic charge between the two. This is precisely what happens in a covalent bond, i.e., buildup of electron charge density between the nuclei. The polarising power of the cation, the polarisability of the anion and the extent of distortion (polarisation) of anion are the factors, which determine the per cent covalent character of the ionic bond.
4.4 THE VALENCE SHELL ELECTRON PAIR REPULSION (VSEPR) THEORY
As already explained, Lewis concept is unable to explain the shapes of molecules. This theory provides a simple procedure to predict the shapes of covalent molecules. Sidgwick
Table 4.5 Dipole Moments of Selected Molecules
| Type of Molecule | Example | Dipole Moment, |
Geometry |
|---|---|---|---|
| Molecule |
1.78 | linear | |
| 1.07 | linear | ||
| 0.79 | linear | ||
| 0.38 | linear | ||
| 0 | linear | ||
| Molecule |
1.85 | bent | |
| 0.95 | bent | ||
| 0 | linear | ||
| Molecule |
1.47 | trigonal-pyramidal | |
| 0.23 | trigonal-pyramidal | ||
| 0 | trigonal-planar | ||
| Molecule |
0 | tetrahedral | |
| 1.04 | tetrahedral | ||
| 0 | tetrahedral |
and Powell in 1940, proposed a simple theory based on the repulsive interactions of the electron pairs in the valence shell of the atoms. It was further developed and redefined by Nyholm and Gillespie (1957).
The main postulates of VSEPR theory are as follows:
- The shape of a molecule depends upon the number of valence shell electron pairs (bonded or nonbonded) around the central atom.
- Pairs of electrons in the valence shell repel one another since their electron clouds are negatively charged.
- These pairs of electrons tend to occupy such positions in space that minimise repulsion and thus maximise distance between them.
- The valence shell is taken as a sphere with the electron pairs localising on the spherical surface at maximum distance from one another.
- A multiple bond is treated as if it is a single electron pair and the two or three electron pairs of a multiple bond are treated as a single super pair.
- Where two or more resonance structures can represent a molecule, the VSEPR model is applicable to any such structure.
The repulsive interaction of electron pairs
decrease in the order:Lone pair (lp) - Lone pair (lp) > Lone pair (lp) - Bond pair (bp) > Bond pair (bp) Bond pair (bp)
Nyholm and Gillespie (1957) refined the VSEPR model by explaining the important difference between the lone pairs and bonding pairs of electrons. While the lone pairs are localised on the central atom, each bonded pair is shared between two atoms. As a result, the lone pair electrons in a molecule occupy more space as compared to the bonding pairs of electrons. This results in greater repulsion between lone pairs of electrons as compared to the lone pair - bond pair and bond pair bond pair repulsions. These repulsion effects result in deviations from idealised shapes and alterations in bond angles in molecules.
For the prediction of geometrical shapes of molecules with the help of VSEPR theory, it is convenient to divide molecules into two categories as (i) molecules in which the central atom has no lone pair and (ii) molecules in which the central atom has one or more lone pairs.
Table 4.6 (page114) shows the arrangement of electron pairs about a central atom A (without any lone pairs) and geometries of some molecules/ions of the type AB. Table 4.7 (page 115) shows shapes of some simple molecules and ions in which the central atom has one or more lone pairs. Table 4.8 (page 116) explains the reasons for the distortions in the geometry of the molecule.
As depicted in Table 4.6, in the compounds of

The VSEPR Theory is able to predict geometry of a large number of molecules, especially the compounds of




4.5 VALENCE BOND THEORY
As we know that Lewis approach helps in writing the structure of molecules but it fails to explain the formation of chemical bond. It also does not give any reason for the difference in bond dissociation enthalpies and bond lengths in molecules like
Similarly the VSEPR theory gives the geometry of simple molecules but theoretically, it does not explain them and also it has limited applications. To overcome these limitations the two important theories based on quantum mechanical principles are introduced. These are valence bond (VB) theory and molecular orbital (MO) theory.
Valence bond theory was introduced by Heitler and London (1927) and developed further by Pauling and others. A discussion of the valence bond theory is based on the knowledge of atomic orbitals, electronic configurations of elements (Units 2), the overlap criteria of atomic orbitals, the hybridization of atomic orbitals and the principles of variation and superposition. A rigorous treatment of the VB theory in terms of these aspects is beyond the scope of this book. Therefore, for the sake of convenience, valence bond theory has been discussed in terms of qualitative and non-mathematical treatment only. To start with, let us consider the formation of hydrogen molecule which is the simplest of all molecules.
Consider two hydrogen atoms A and B approaching each other having nuclei
Attractive forces arise between:
(i) nucleus of one atom and its own electron that is
Similarly repulsive forces arise between
(i) electrons of two atoms like
(ii) nuclei of two atoms
Attractive forces tend to bring the two atoms close to each other whereas repulsive forces tend to push them apart (Fig. 4.7).

Experimentally it has been found that the magnitude of new attractive force is more than the new repulsive forces. As a result, two atoms approach each other and potential energy decreases. Ultimately a stage is reached where the net force of attraction balances the force of repulsion and system acquires minimum energy. At this stage two hydrogen atoms are said to be bonded together to form a stable molecule having the bond length of
Since the energy gets released when the bond is formed between two hydrogen atoms, the hydrogen molecule is more stable than that of isolated hydrogen atoms. The energy so released is called as bond enthalpy, which is corresponding to minimum in the curve depicted in Fig. 4.8. Conversely,

4.5.1 Orbital Overlap Concept
In the formation of hydrogen molecule, there is a minimum energy state when two hydrogen atoms are so near that their atomic orbitals undergo partial interpenetration. This partial merging of atomic orbitals is called overlapping of atomic orbitals which results in the pairing of electrons. The extent of overlap decides the strength of a covalent bond. In general, greater the overlap the stronger is the bond formed between two atoms. Therefore, according to orbital overlap concept, the formation of a covalent bond between two atoms results by pairing of electrons present in the valence shell having opposite spins.
4.5.2 Directional Properties of Bonds
As we have already seen, the covalent bond is formed by overlapping of atomic orbitals. The molecule of hydrogen is formed due to the overlap of
In case of polyatomic molecules like
The valence bond theory explains the shape, the formation and directional properties of bonds in polyatomic molecules like
4.5.3 Overlapping of Atomic Orbitals
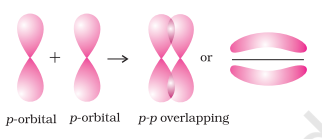
When orbitals of two atoms come close to form bond, their overlap may be positive, negative or zero depending upon the sign (phase) and direction of orientation of amplitude of orbital wave function in space (Fig. 4.9). Positive and negative sign on boundary surface diagrams in the Fig. 4.9 show the sign (phase) of orbital wave function and are not related to charge. Orbitals forming bond should have same sign (phase) and orientation in space. This is called positive overlap. Various overlaps of
The criterion of overlap, as the main factor for the formation of covalent bonds applies uniformly to the homonuclear/heteronuclear diatomic molecules and polyatomic molecules. We know that the shapes of
Let us first consider the

hydrogen. The four atomic orbitals of carbon, each with an unpaired electron can overlap with the
4.5.4 Types of Overlapping and Nature of Covalent Bonds
The covalent bond may be classified into two types depending upon the types of overlapping:
(i) Sigma(
(i) Sigma(



(ii) pi(
4.5.5 Strength of Sigma and pi Bonds
Basically the strength of a bond depends upon the extent of overlapping. In case of sigma bond, the overlapping of orbitals takes place to a larger extent. Hence, it is stronger as compared to the pi bond where the extent of overlapping occurs to a smaller extent. Further, it is important to note that in the formation of multiple bonds between two atoms of a molecule, pi bond(s) is formed in addition to a sigma bond.
4.6 HYBRIDISATION
In order to explain the characteristic geometrical shapes of polyatomic molecules like
Salient features of hybridisation: The main features of hybridisation are as under :
1. The number of hybrid orbitals is equal to the number of the atomic orbitals that get hybridised.
2. The hybridised orbitals are always equivalent in energy and shape.
3. The hybrid orbitals are more effective in forming stable bonds than the pure atomic orbitals.
4. These hybrid orbitals are directed in space in some preferred direction to have minimum repulsion between electron pairs and thus a stable arrangement. Therefore, the type of hybridisation indicates the geometry of the molecules.
Important conditions for hybridisation
(i) The orbitals present in the valence shell of the atom are hybridised.
(ii) The orbitals undergoing hybridisation should have almost equal energy.
(iii) Promotion of electron is not essential condition prior to hybridisation.
(iv) It is not necessary that only half filled orbitals participate in hybridisation. In some cases, even filled orbitals of valence shell take part in hybridisation.
4.6.1 Types of Hybridisation
There are various types of hybridisation involving
(I) sp hybridisation: This type of hybridisation involves the mixing of one
The two
Example of molecule having

(II)

a result boron has three unpaired electrons. These three orbitals (one
(III)

The structure of

In case of

4.6.2 Other Examples of
sp
Thus, in ethene molecule, the carboncarbon bond consists of one

sp Hybridisation in
One

4.6.3 Hybridisation of Elements involving d Orbitals
The elements present in the third period contain
The important hybridisation schemes involving

(i) Formation of
The ground state and the excited state outer electronic configurations of phosphorus

Now the five orbitals (i.e., one

It should be noted that all the bond angles in trigonal bipyramidal geometry are not equivalent. In
(ii) Formation of


4.7 MOLECULAR ORBITAL THEORY
Molecular orbital (MO) theory was developed by F. Hund and R.S. Mulliken in 1932. The salient features of this theory are :
(i) The electrons in a molecule are present in the various molecular orbitals as the electrons of atoms are present in the various atomic orbitals.
(ii) The atomic orbitals of comparable energies and proper symmetry combine to form molecular orbitals.
(iii) While an electron in an atomic orbital is influenced by one nucleus, in a molecular orbital it is influenced by two or more nuclei depending upon the number of atoms in the molecule. Thus, an atomic orbital is monocentric while a molecular orbital is polycentric.
(iv) The number of molecular orbital formed is equal to the number of combining atomic orbitals. When two atomic orbitals combine, two molecular orbitals are formed. One is known as bonding molecular orbital while the other is called antibonding molecular orbital.
(v) The bonding molecular orbital has lower energy and hence greater stability than the corresponding antibonding molecular orbital.
(vi) Just as the electron probability distribution around a nucleus in an atom is given by an atomic orbital, the electron probability distribution around a group of nuclei in a molecule is given by a molecular orbital.
(vii) The molecular orbitals like atomic orbitals are filled in accordance with the aufbau principle obeying the Pauli’s exclusion principle and the Hund’s rule.
4.7.1 Formation of Molecular Orbitals Linear Combination of Atomic Orbitals (LCAO)
According to wave mechanics, the atomic orbitals can be expressed by wave functions (
Let us apply this method to the homonuclear diatomic hydrogen molecule. Consider the hydrogen molecule consisting of two atoms A and B. Each hydrogen atom in the ground state has one electron in
Therefore, the two molecular orbitals
The molecular orbital

Qualitatively, the formation of molecular orbitals can be understood in terms of the constructive or destructive interference of the electron waves of the combining atoms. In the formation of bonding molecular orbital, the two electron waves of the bonding atoms reinforce each other due to constructive interference while in the formation of antibonding molecular orbital, the electron waves cancel each other due to destructive interference. As a result, the electron density in a bonding molecular orbital is located between the nuclei of the bonded atoms because of which the repulsion between the nuclei is very less while in case of an antibonding molecular orbital, most of the electron density is located away from the space between the nuclei. Infact, there is a nodal plane (on which the electron density is zero) between the nuclei and hence the repulsion between the nuclei is high. Electrons placed in a bonding molecular orbital tend to hold the nuclei together and stabilise a molecule. Therefore, a bonding molecular orbital always possesses lower energy than either of the atomic orbitals that have combined to form it. In contrast, the electrons placed in the antibonding molecular orbital destabilise the molecule. This is because the mutual repulsion of the electrons in this orbital is more than the attraction between the electrons and the nuclei, which causes a net increase in energy.
It may be noted that the energy of the antibonding orbital is raised above the energy of the parent atomic orbitals that have combined and the energy of the bonding orbital has been lowered than the parent orbitals. The total energy of two molecular orbitals, however, remains the same as that of two original atomic orbitals.
4.7.2 Conditions for the Combination of Atomic Orbitals
The linear combination of atomic orbitals to form molecular orbitals takes place only if the following conditions are satisfied:
1. The combining atomic orbitals must have the same or nearly the same energy. This means that
2. The combining atomic orbitals must have the same symmetry about the molecular axis. By convention
3. The combining atomic orbitals must overlap to the maximum extent. Greater the extent of overlap, the greater will be the electron-density between the nuclei of a molecular orbital.
4.7.3 Types of Molecular Orbitals
Molecular orbitals of diatomic molecules are designated as
In this nomenclature, the sigma
Molecular orbitals obtained from
4.7.4 Energy Level Diagram for Molecular Orbitals
We have seen that

on two atoms) give rise to the following eight molecular orbitals:
The energy levels of these molecular orbitals have been determined experimentally from spectroscopic data for homonuclear diatomic molecules of second row elements of the periodic table. The increasing order of
energies of various molecular orbitals for
However, this sequence of energy levels of molecular orbitals is not correct for the remaining molecules
The important characteristic feature of this order is that the energy of
4.7.5 Electronic Configuration and Molecular Behaviour
The distribution of electrons among various molecular orbitals is called the electronic configuration of the molecule. From the electronic configuration of the molecule, it is possible to get important information about the molecule as discussed below.
Stability of Molecules: If
(i) the molecule is stable if
(ii) the molecule is unstable if
In (i) more bonding orbitals are occupied and so the bonding influence is stronger and a stable molecule results. In (ii) the antibonding influence is stronger and therefore the molecule is unstable.
Bond order
Bond order (b.o.) is defined as one half the difference between the number of electrons present in the bonding and the antibonding orbitals i.e.,
Bond order (b.o.)
The rules discussed above regarding the stability of the molecule can be restated in terms of bond order as follows: A positive bond order (i.e.,
Nature of the bond
Integral bond order values of 1,2 or 3 correspond to single, double or triple bonds respectively as studied in the classical concept.
Bond-length
The bond order between two atoms in a molecule may be taken as an approximate measure of the bond length. The bond length decreases as bond order increases.
Magnetic nature
If all the molecular orbitals in a molecule are doubly occupied, the substance is diamagnetic (repelled by magnetic field). However if one or more molecular orbitals are singly occupied it is paramagnetic (attracted by magnetic field), e.g.,
4.8 BONDING IN SOME HOMONUCLEAR DIATOMIC MOLECULES
In this section we shall discuss bonding in some homonuclear diatomic molecules.
1. Hydrogen molecule
The bond order of
This means that the two hydrogen atoms are bonded together by a single covalent bond. The bond dissociation energy of hydrogen molecule has been found to be
2. Helium molecule
Bond order of
Similarly, it can be shown that
3. Lithium molecule
The above configuration is also written as
From the electronic configuration of
4. Carbon molecule
or
The bond order of
5. Oxygen molecule
From the electronic configuration of
Bond order
So in oxygen molecule, atoms are held by a double bond. Moreover, it may be noted that it contains two unpaired electrons in
Similarly, the electronic configurations of other homonuclear diatomic molecules of the second row of the periodic table can be written. In Fig. 4.21 are given the molecular orbital occupancy and molecular properties for

4.9 HYDROGEN BONDING
Nitrogen, oxygen and fluorine are the highly electronegative elements. When they are attached to a hydrogen atom to form covalent bond, the electrons of the covalent bond are shifted towards the more electronegative atom. This partially positively charged hydrogen atom forms a bond with the other more electronegative atom. This bond is known as hydrogen bond and is weaker than the covalent bond. For example, in HF molecule, the hydrogen bond exists between hydrogen atom of one molecule and fluorine atom of another molecule as depicted below :
Here, hydrogen bond acts as a bridge between two atoms which holds one atom by covalent bond and the other by hydrogen bond.
Hydrogen bond is represented by a dotted line (
4.9.1 Cause of Formation of Hydrogen Bond
When hydrogen is bonded to strongly electronegative element ’
The magnitude of
4.9.2 Types of
There are two types of
(i) Intermolecular hydrogen bond
(ii) Intramolecular hydrogen bond
(1) Intermolecular hydrogen bond : It is formed between two different molecules of the same or different compounds. For example,
(2) Intramolecular hydrogen bond : It is formed when hydrogen atom is in between the two highly electronegative

Summary
Kössel’s first insight into the mechanism of formation of electropositive and electronegative ions related the process to the attainment of noble gas configurations by the respective ions. Electrostatic attraction between ions is the cause for their stability. This gives the concept of electrovalency.
The first description of covalent bonding was provided by Lewis in terms of the sharing of electron pairs between atoms and he related the process to the attainment of noble gas configurations by reacting atoms as a result of sharing of electrons. The Lewis dot symbols show the number of valence electrons of the atoms of a given element and Lewis dot structures show pictorial representations of bonding in molecules.
An ionic compound is pictured as a three-dimensional aggregation of positive and negative ions in an ordered arrangement called the crystal lattice. In a crystalline solid there is a charge balance between the positive and negative ions. The crystal lattice is stabilized by the enthalpy of lattice formation.
While a single covalent bond is formed by sharing of an electron pair between two atoms, multiple bonds result from the sharing of two or three electron pairs. Some bonded atoms have additional pairs of electrons not involved in bonding. These are called lone-pairs of electrons. A Lewis dot structure shows the arrangement of bonded pairs and lone pairs around each atom in a molecule. Important parameters, associated with chemical bonds, like: bond length, bond angle, bond enthalpy, bond order and bond polarity have significant effect on the properties of compounds.
A number of molecules and polyatomic ions cannot be described accurately by a single Lewis structure and a number of descriptions (representations) based on the same skeletal structure are written and these taken together represent the molecule or ion. This is a very important and extremely useful concept called resonance. The contributing structures or canonical forms taken together constitute the resonance hybrid which represents the molecule or ion.
The VSEPR model used for predicting the geometrical shapes of molecules is based on the assumption that electron pairs repel each other and, therefore, tend to remain as far apart as possible. According to this model, molecular geometry is determined by repulsions between lone pairs and lone pairs; lone pairs and bonding pairs and bonding pairs and bonding pairs. The order of these repulsions being : lp-lp
The valence bond (VB) approach to covalent bonding is basically concerned with the energetics of covalent bond formation about which the Lewis and VSEPR models are silent. Basically the VB theory discusses bond formation in terms of overlap of orbitals. For example the formation of the
For explaining the characteristic shapes of polyatomic molecules Pauling introduced the concept of hybridisation of atomic orbitals.
The molecular orbital (MO) theory describes bonding in terms of the combination and arrangment of atomic orbitals to form molecular orbitals that are associated with the molecule as a whole. The number of molecular orbitals are always equal to the number of atomic orbitals from which they are formed. Bonding molecular orbitals increase electron density between the nuclei and are lower in energy than the individual atomic orbitals. Antibonding molecular orbitals have a region of zero electron density between the nuclei and have more energy than the individual atomic orbitals.
The electronic configuration of the molecules is written by filling electrons in the molecular orbitals in the order of increasing energy levels. As in the case of atoms, the Pauli exclusion principle and Hund’s rule are applicable for the filling of molecular orbitals. Molecules are said to be stable if the number of elctrons in bonding molecular orbitals is greater than that in antibonding molecular orbitals.
Hydrogen bond is formed when a hydrogen atom finds itself between two highly electronegative atoms such as F, O and N. It may be intermolecular (existing between two or more molecules of the same or different substances) or intramolecular (present within the same molecule). Hydrogen bonds have a powerful effect on the structure and properties of many compounds.





